
The Bureaucracy Blocking the Chance at a Cure
How early-stage clinical trials became unnecessarily expensive and inefficient—and how we can fix them, inspired by a recent story about a dog treated for cancer.

A story about Paul Conyngham, an AI entrepreneur from Sydney who treated his dog Rosie’s cancer with a personalized mRNA vaccine, has been circulating on X since yesterday. What makes the story inspiring is the initiative the owner showed: he used AI to teach himself about how a personalized vaccine could work, designed much of the process himself and approached top researchers to take it forward.
Whether the treatment itself was curative and how much of an improvement it represents over state-of-the art is not the main focus of this essay. Others have already debated that question at length, and I recommend following their discussions1.
What interests me instead is the bureaucratic absurdity the dog’s owner encountered while trying to pursue the treatment. He described the long and frustrating process required simply to test the drug in his dog: “The red tape was actually harder than the vaccine creation, and I was trying to get an Australian ethics approval and run a dog trial on Rosie. It took me three months, putting two hours aside every single night, just typing the 100 page document.” Even in a small and urgent case, where the owner was fully willing to fund the treatment himself, the effort was slowed by layers of procedure.
Of course, this kind of red tape is not confined to Australia, nor to veterinary medicine. In fact, in the US, the red tape is even worse, at least for in-human trials.
In the United States, GitLab co-founder Sid Sijbrandij found himself in a similar position after the relapse of his osteosarcoma. When the ordinary doors of medicine closed, he entered what he called “founder mode on his cancer.” Like many entrepreneurs confronted with a difficult problem, he began trying to build his own path forward by self-funding his exploration of experimental therapies.
Even then, he ran into the same maze of regulatory and institutional barriers that not only delayed him, but also unnecessarily raised the price of his experimental therapies. These are obstacles that only someone with extraordinary resources could hope to navigate, often by assembling an entire team to deal with them and navigate the opacity. In the end, Sijbrandij prevailed: he has been relapse free since 2025, after doctors had told him he was at the end of his options.
Around the same time, writer Jake Seliger faced a similar situation while battling advanced throat cancer. Like Sid Sijbrandij, he was willing to try anything that might help. The difference was that Seliger was not a billionaire. He could not hire a team to navigate the system on his behalf, and he struggled even to enroll in the clinical trials that might have offered him a chance.
A system originally conceived to safeguard patients has gradually produced a strange and troubling outcome: the mere chance of survival is effectively reserved for the very few who possess the means to assemble an army of experts capable of navigating its labyrinthine procedures.
What makes these stories particularly frustrating is that we already know clinical trials — especially small, early-stage ones like the ones Sijbrandij enrolled in for himself— can be conducted far more cheaply and with far less bureaucracy than is currently required. Ironically, the original article cites Australia as a bad example, yet clinical trials there are conducted 2.5–3× cheaper and faster than in the U.S., at least for human trials, without any increase in safety events—a genuine free lunch.
Removing unnecessary barriers has long been important. That is why I co-founded the Clinical Trial Abundance initiative in 2024, a policy effort aimed at increasing both the number and efficiency of in-human drug trials and have consistently argued about the importance of making this crucial but often neglected part of the drug discovery process more efficient.
Since then, the issue has only become more urgent with the rise of AI. One of the central promises of the AI revolution is that it will accelerate medical progress. Organizations such as the OpenAI Foundation list curing disease as a core goal, and researchers like Dario Amodei of Anthropic have argued that AI could dramatically speed up biomedical innovation. But, as I have written before in response to an interview between Dario and Dwarkesh Patel, AI will not automatically accelerate a key bottleneck in making these dreams a reality: clinical trials. Conyngham’s observation that navigating the red tape to start a trial for his dog took longer than designing the drug itself only underscores the point.
Clinical trials themselves vary widely. At one end are small, bespoke trials involving one or a few patients testing highly experimental therapies—like the treatment in the Australian dog story or the experimental therapy Sijbrandij pursued. At the other end are large-scale trials involving thousands of participants, designed to confirm earlier findings and support regulatory approval.
Different types of trials require different reforms. In this essay, I will focus on the former: small, exploratory trials, which will be called early-stage small n trials for the purpose of this essay. These are often the fastest way to test promising ideas in humans and learn from them. They represent our best chance at a meaningful “right-to-try,” form the top of the funnel that generates proof-of-concept evidence, and may be the only viable path for personalized medicine and treatments for ultra-rare diseases. Understanding why these trials have been made unnecessarily difficult—and how we might change that—is essential if medical innovation is to keep pace with our growing ability to design new therapies.
The real problem: We make early stage trials prohibitively hard
When the story first circulated on X, many people interpreted it as evidence that a cure already exists but simply hasn’t been used due to bureaucracy. That isn’t quite true, as I explained.
The type of mRNA vaccine that the owner pursued looks promising, but he did not know a priori whether it worked or not, as it had not been tested before. So it was not a cure, but “a chance at a cure”. I hesitate to call it an “experimental treatment”, since this term evokes fears of potential safety issues while we generally can predict safety quite well now. The inaccuracy of whether this was a cure or not, however, does not make the story of the bureaucratic red tape that Conyngham encountered any less infuriating. More and more promising treatments are accumulating in the pipeline, fueled by an explosion of new therapeutic modalities, ranging from mRNA to better peptides and more recently, by AI. Yet we are not taking full advantage of them.
To better understand these points, it is helpful to briefly outline the clinical development process—the sequence of in-human trials through which a promising scientific idea is gradually translated into a therapy.
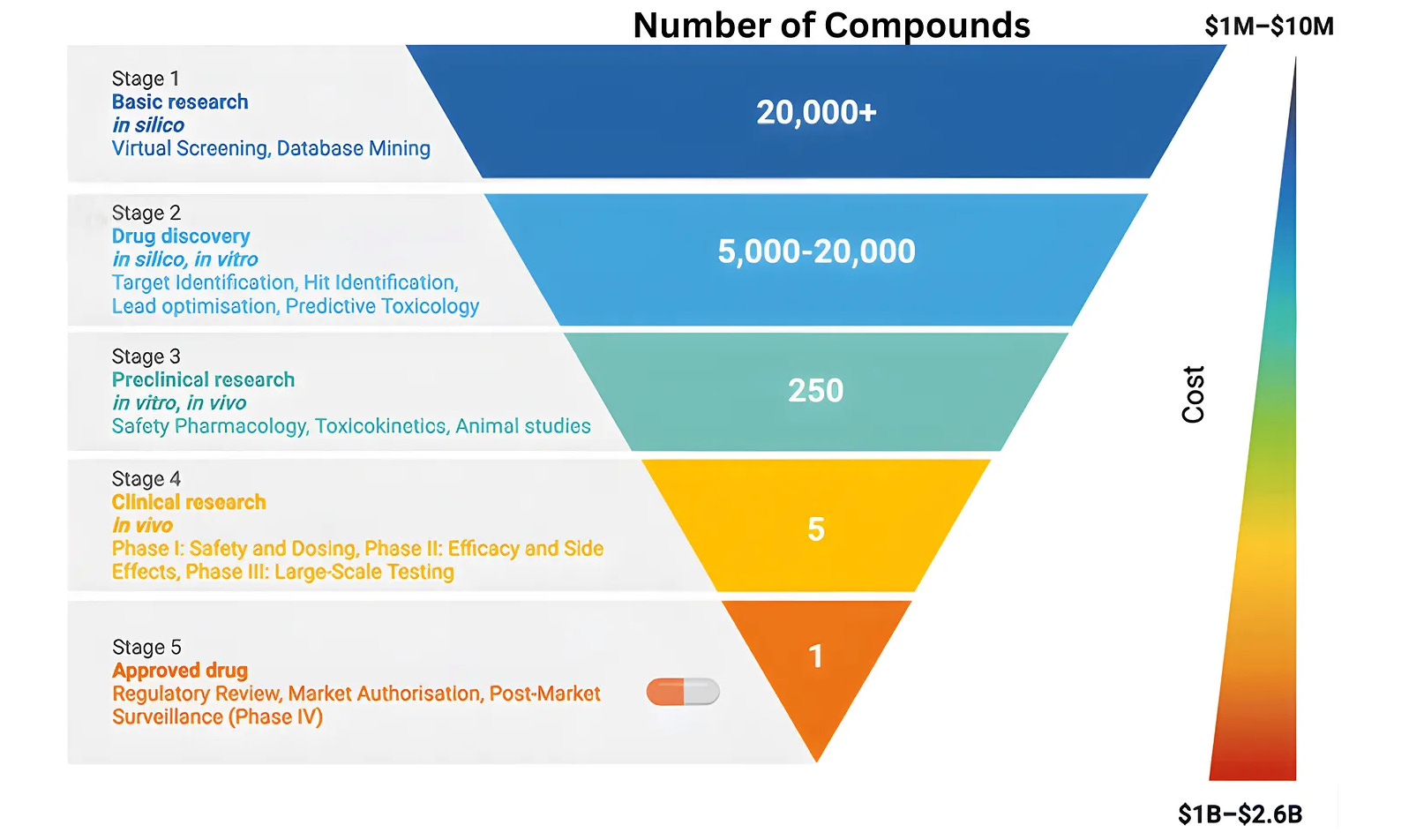
Drug development is often described as a funnel: many ideas enter at the top, but only a few become approved treatments. Early human studies, known as Phase I trials, sit at the entrance of this process. They involve small numbers of patients and are designed to quickly test whether a new therapy is safe and shows early signs of effectiveness.
If the results look promising, the therapy moves to larger and more complex studies, including Phase III trials that enroll large numbers of patients to confirm whether the treatment truly works. Most people gain access to new therapies only after these large randomized trials are completed.
On average, moving from a promising idea to Phase III results takes seven to ten years and costs roughly $1.2 billion. Accelerated approval pathways in areas such as cancer or rare diseases can shorten this timeline by relying on surrogate endpoints, but the process remains slow. As a result, many discoveries that make headlines today will take close to a decade before they become treatments that patients can widely access.
Part of this delay is unavoidable. Observing how a drug affects the human body simply takes time. But much of it is not. Layers of unnecessary bureaucracy, regulatory opacity, and rising trial costs add years to the process without clearly improving patient safety, which is why I started Clinical Trial Abundance.
Allowing a higher volume of small-n early stage trials, the focus of this essay, is a rare “win-win” for both public health and scientific progress. For patients, it transforms a terminal diagnosis from a closed door into a “chance at a cure,” providing legal, supervised access to cutting-edge medicine that currently sits idle in labs. For researchers and society, it unclogs the drug discovery funnel; by lowering the barrier to entry for new ideas, we ensure that the next generation of mRNA, peptide and AI-driven therapies are tested in humans years sooner, ultimately accelerating the arrival of universal cures for everyone.
Next, I will explain why making it easier to run these early stage trials matters.
First, from a patient perspective, they often provide the closest practical equivalent to a right-to-try. In theory, right-to-try laws allow patients with serious illnesses to access treatments that have not yet been confirmed in large randomized Phase III trials. In practice, these pathways rarely function as intended. Pharmaceutical companies are often reluctant to provide experimental drugs outside formal trials, and treatments typically must have already passed Phase I testing. As a result, very few patients gain access through these mechanisms. Early-stage trials offer a more workable alternative. They allow experimental therapies to be tested in structured clinical environments—often in academic settings or academia–industry collaborations—where patients can be monitored and meaningful data can be collected.
Second, early-stage small-n trials are essential for personalized medicine and the treatment of ultra-rare diseases. Many emerging therapies—such as personalized cancer vaccines, gene therapies, and other individualized interventions—do not fit easily into the traditional model of large randomized trials involving thousands of participants. By their nature, these treatments target very small patient populations and often require flexible, adaptive clinical designs.
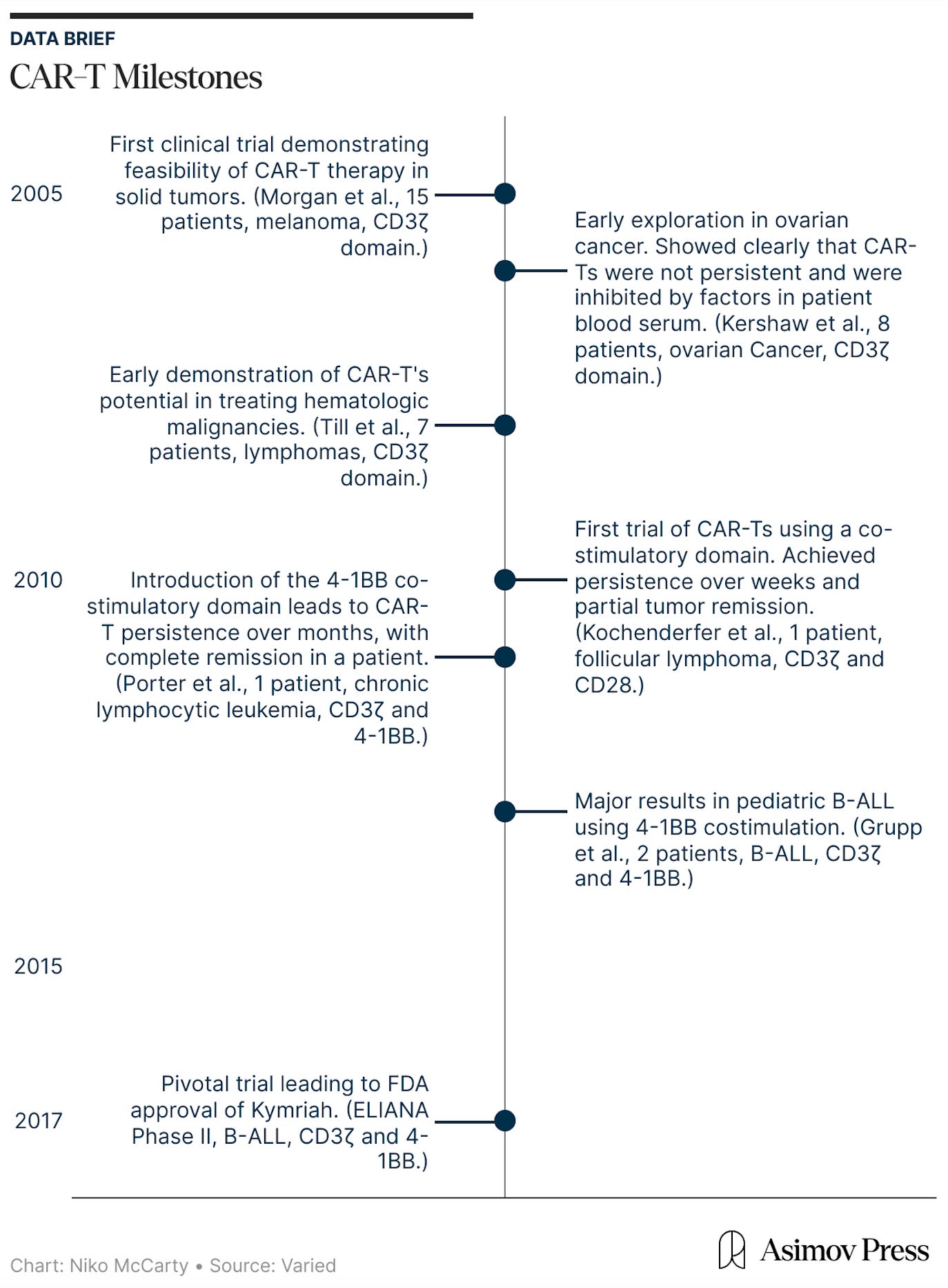
From a societal perspective, these trials play a crucial learning role. As I argued in my earlier essay Clinic-in-the-Loop, early-stage trials are not simply regulatory checkpoints on the path to approval. They are part of the discovery process itself, creating a feedback loop between laboratory hypotheses and human biology. Later-stage studies, particularly Phase III trials, are designed mainly for validation: they test whether a treatment works under defined conditions and produce the evidence needed for approval.
Early-stage trials, by contrast, are oriented toward learning. Conducted with small patient groups and often using exploratory designs, they allow researchers to observe how a therapy behaves in the human body and how the disease responds. In this way, they close the gap between theory and real-world biology. In the Clinic-in-the-Loop essay, I explain how these trials were crucial to the discovery of Kymriah, the first curative cell therapy for blood cancer.
Lastly, these trials play an important role in maintaining U.S. leadership in biotechnology and, over the long run, in safeguarding biosecurity. In recent years, China has been advancing rapidly in biotechnology, in part because it is easier to run early-stage clinical studies there. These studies, often referred to as Investigator-Initiated Trials (IITs), allow researchers to test new ideas quickly and at relatively low cost.
As a result, more U.S. biotech firms are beginning to move parts of their clinical development to China. A Time magazine headline from May 2025 captures what many industry experts have been warning for years: “The US can’t afford to lose the biotech race with China.” This is no longer a hypothetical concern. Data is backing this fear up: US early-stage funding is deteriorating: dropping from $2.6 billion in Q1 to just $900 million in Q2 2025 — the lowest level in five quarters. If this trend continues, it could gradually shift the center of gravity for biomedical innovation abroad. In the long run, this poses risks not only to U.S. biotech competitiveness but also to biosecurity, much like the earlier offshoring of manufacturing supply chains created strategic vulnerabilities.
In the long run, we may need to rethink the entire sequence of requirements for drug approval, especially as personalized medicine becomes more common. For now, however, it is worth focusing on the unnecessary barriers that limit the expansion of early-stage trials to a larger group of patients.
Regulatory Barriers to Early-Stage Clinical Trials
A true technologist at heart, Sid Sijbrandij documented every medical interaction in extensive health notes, gathered and stored raw results from scans, blood tests, genomic sequencing, and tissue analysis, and assembled a team of concierge doctors, researchers, advisors, and operators to help manage and interpret everything and help him design his own therapies. You can read a summary of his journey here and here.
Although his determination was strong, he encountered numerous obstacles. One of them were the Institutional Review Boards (IRBs), committees responsible for reviewing the ethical aspects of clinical studies. In practice, however, the IRB process significantly delayed his efforts. As Sid explained in a Century of Bio article, they can function as “a ‘vetocracy’ where one member of the board can block treatment based on even the smallest concern.”
This situation is difficult to justify: here was someone with advanced cancer who was willing to self-fund the treatment and accept the risks, yet was still prevented from proceeding. It’s as if the system would rather have you dead than risk one imperfect overly long consent form.
In one of my policy proposals for Institute for Progress, part of the Clinical Trial Abundance initiative, I trace the history of IRBs and examine some of the dysfunctions and unintended consequences of the current system.
IRBs were created to safeguard the ethical treatment of human research participants. Over time, however, many institutional IRBs have drifted away from this core mission. What began as collegial ethical oversight has gradually evolved into a bureaucratic compliance system that prioritizes documentation, procedure, and institutional risk management over substantive ethical judgment.
Because universities typically require researchers to use their own institutional IRB, investigators often have no practical alternative when reviews become arbitrary or excessively slow. At the same time, the true costs of IRB administration are largely hidden within indirect grant funding, insulating IRBs from accountability and obscuring the financial impact of inefficiency. The result is a fragmented oversight landscape that imposes significant administrative burdens and delays on research while offering little evidence that these additional procedural layers meaningfully improve protections for human subjects.
What makes this situation particularly frustrating is that better models already exist. Independent IRBs regularly demonstrate faster review timelines and comparable or lower costs than many institutional IRBs, while holding the highest standards of ethical accreditation. Despite operating under the same federal regulatory framework, these boards compete on service, efficiency, and expertise. Yet investigators at hospitals and academic medical centers are typically prevented from accessing them by internal institutional policies. There is little justification for a system in which academic researchers are effectively locked into a single IRB provider when qualified, federally compliant alternatives are readily available.
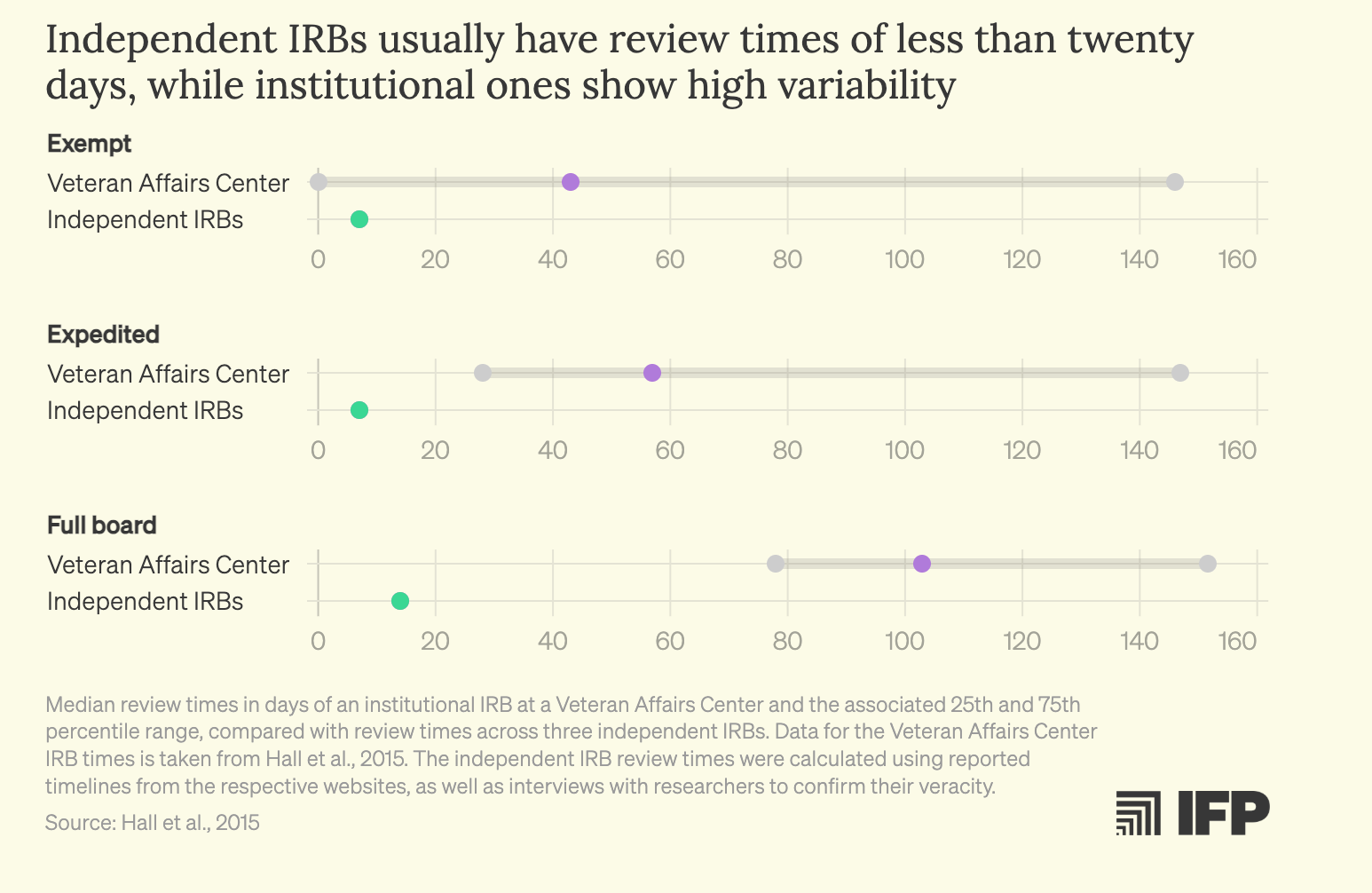
One would expect they are more expensive but it turns out… they are not!
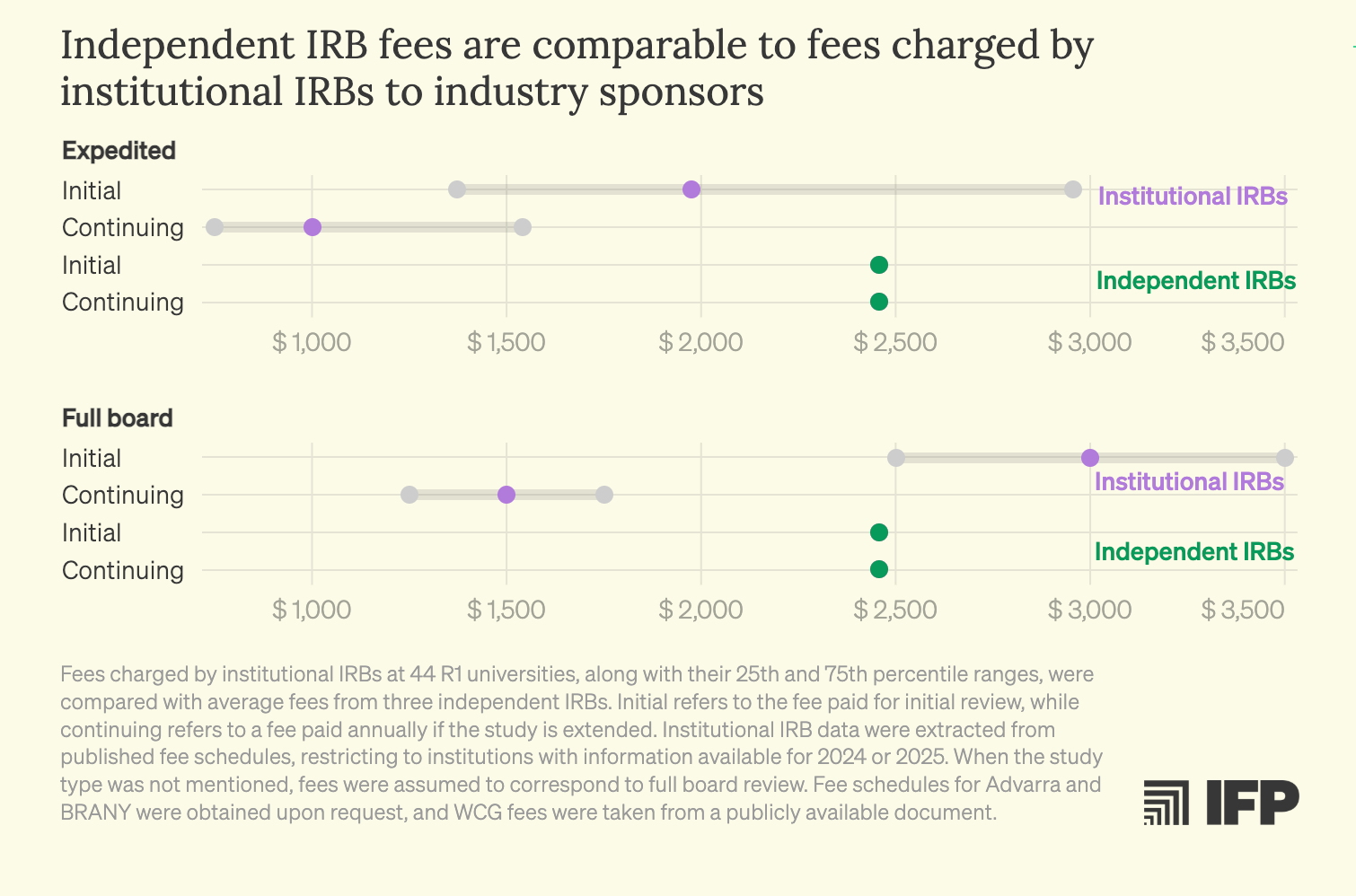
While IRBs are one example of frustrating delay in time, there are also regulatory requirements that make experimental therapies unnecessarily costly.
One of the largest cost drivers in early-stage trials comes from manufacturing regulations, specifically the requirements around Chemistry, Manufacturing, and Controls (CMC) and Good Manufacturing Practice (GMP) standards. CMC refers to the detailed process and validation of how a drug is produced, including the materials used, the production process, quality controls, and stability testing. These requirements are essential when a therapy is being produced at scale for widespread use. However, in the context of very small early-stage trials involving only a handful of patients, the same standards are often applied even though the risks and scale are entirely different.
GMP is the regulatory framework that governs how drugs must be manufactured to ensure safety, consistency, and quality. Full GMP compliance involves validated facilities, extensive documentation, batch testing, environmental monitoring, and strict process controls. These requirements make sense for large commercial manufacturing, where thousands or millions of patients may ultimately receive the product. But they are extremely expensive and time-consuming to implement, often adding millions of dollars and many months of delay before a therapy can even be tested in humans.
For small early-stage trials, a more proportionate approach is possible. Some regulatory systems already allow what could be called “GMP-light” manufacturing—production that follows core safety and quality principles but does not require the full set of industrial-scale validation steps. Under such a model, therapies can be manufactured in controlled research facilities using well-understood materials and processes, while still maintaining safeguards against contamination or gross quality failures. The key difference is that the level of documentation and process validation is scaled to the size and exploratory purpose of the trial.
How large could the decreases in cost be?
Experience in Australia shows that a lighter approach to Good Manufacturing Practice (GMP) requirements can substantially reduce costs. Clinical manufacturing conducted under Australia’s framework is roughly 2.5x cheaper for this stage of the process alone. Importantly, Australia has operated under this model for about three decades without any observed increase in safety events. This suggests that a 2.5x reduction should be seen as a conservative lower bound on the potential savings from regulatory reform.
Lower CMC requirements also reduce costs indirectly. By allowing much faster entry into the clinic, they enable biotechnology companies to obtain proof-of-concept earlier. This matters because the financial burn rate of a clinical-stage biotech is extremely high—often around $25 million per year. Delays in reaching proof-of-concept can therefore be existential for smaller firms. In practice, prolonged timelines can exhaust capital before meaningful clinical results are obtained.
For this reason, in recent years many smaller biotechs have shifted early clinical operations from the United States to Australia. This is bad in two important ways. Firstly, it deprives U.S. patients from access to state-of-the art therapies. Secondly, it threatens to weaken the biotech sector in the U.S as a whole. In addition, Australia itself is getting “crowded.” At a population of slightly less than 30 million, there are only so many trials it can support.
The direct 2.5x reduction in manufacturing costs is likely only the beginning. If regulatory reforms were combined with modern technologies for final product quality control — such as improved analytical assays and standardized validation platforms — experts I interviewed suggest that manufacturing costs could plausibly fall by 5–10× overall. Such improvements would not only lower the cost of individual trials but could also significantly expand the number of therapies that make it into clinical testing in the first place.
Lowering costs would effectively expand access to early-stage clinical trials. Today, the high cost of manufacturing and running these trials means that only a small number of patients can participate. As an academic immuno-oncologist working in cell therapy at a prestigious U.S. academic institution —who asked to remain anonymous — told me, this often leads to heartbreaking choices. Because the size of academic grants only allows treatment for a handful of patients, he is forced to decide which patients receive the therapy and which do not.
Solutions
I am currently working with Institute for Progress and OneDaySooner on concrete policy proposals for making early-stage clinical trials faster and cheaper by drafting and implementation frameworks that could realistically be adopted.
This work requires navigating significant opacity in the current regulatory and operational ecosystem. As I have written before here and here, the regulatory process is completely opaque and locked behind trade secret protections — which is a problem in and of itself.
Many of the key processes involved in clinical trial manufacturing and quality control are handled by specialized third-party vendors that operate under strict nondisclosure agreements (NDAs). As a result, even basic information about timelines, costs, and regulatory interpretation is fragmented and difficult to access. Despite these constraints, several promising reform directions are emerging.
Here I summarize some of the most promising proposals:
Improve ethics review by allowing investigator choice of IRB.
Granting academic investigators the freedom to choose among accredited IRBs would better align incentives across the IRB ecosystem, introducing competition, cost transparency, and accountability. This could be achieved with two policy shifts:
(A) Guaranteeing the right of federally funded investigators to select an external IRB as the IRB of record. This would be coupled with robust non-retaliation rules to protect researchers who choose an external IRB, and the elimination of duplicative internal review once an external IRB issues a determination. By allowing researchers to choose among compliant IRBs, and by making those options more visible, market discipline and accountability would be reintroduced into the oversight process.
(B) Prohibiting institutions from funding IRB operations through indirect costs. Decoupling IRBs from Facilities and Administrative costs will make prices more transparent, enabling researchers to make more informed choices, and encouraging competition in terms of speed, clarity, and rigor.
For more details on this, you can check the full proposal here.
Implement a notification pathway for early-stage trials.
A regulatory structure modeled after Australia’s Clinical Trial Notification (CTN) framework offers a concrete example of the kind of policy push that could speed up these types of trials. There, most early-phase trials proceed after approval by a Human Research Ethics Committee (HREC), with notification rather than pre-approval by the regulator. The regulator retains inspection powers and the authority to halt unsafe studies, but does not duplicate the scientific review already conducted by the clinician-scientists and toxicologists embedded in HRECs. The result is that clinical trial sites can begin giving drugs to patients much sooner (about two times faster than in the United States, according to informal interviews with industry leaders).
In the United States, by contrast, Phase I trials typically require submission of an Investigational New Drug (IND) application to the U.S. Food and Drug Administration before initiation. This dual review — by both an IRB and the federal regulator — creates redundancy that lengthens the feedback loop. A CTN-like model for Phase I trials could preserve safety oversight while shifting scientific and toxicological reviews to accredited, transparently governed IRBs with expanded expertise. The FDA would retain the power to inspect, impose clinical holds, and intervene in high-risk cases, such as for novel gene therapies. But for the majority of small-molecule first-in-human studies, the default could be notification rather than permission.
Modify statutory language to enable more flexible GMP requirements.
Although in the U.S., Phase I investigational drugs are technically exempt from the detailed manufacturing rules in 21 CFR Part 211, the exemption offers far less practical relief than it appears. Part 211 is the section of the U.S. Code of Federal Regulations that specifies the current Good Manufacturing Practice (cGMP) requirements for finished pharmaceutical products. These rules govern how drugs are manufactured at a commercial scale and cover areas such as facility design, equipment validation, environmental controls, quality control systems, batch testing, stability studies, and detailed recordkeeping to ensure consistency and safety across large production runs.
The regulation (21 CFR §210.2(c)) states that Phase I drugs do not need to comply with Part 211, but it simultaneously requires that they still satisfy the underlying statutory requirement that drugs be manufactured according to cGMP under 21 U.S.C. §351(a)(2)(B) which states that drugs not manufactured under GMP are “adulterated.”Being classified as adulterated carries serious consequences. Because the statute does not clearly specify what level of GMP is appropriate for a small exploratory trial versus a commercial product manufactured at scale and what adulterated, manufacturers typically adopt the safest interpretation and follow nearly the full set of commercial GMP standards.
In practice, this ambiguity creates a de facto GMP floor, where companies default to the most expensive manufacturing standards simply to avoid regulatory risk. Clarifying or explicitly scaling GMP expectations for early-stage trials would allow proportionate standards. However, a key step is first modifying U.S.C. §351(a)(2)(B).
These would be just the beginnings.
The U.S. can do better than Australia. One option is to relax certain CMC (Chemistry, Manufacturing, and Controls) requirements and shift some verification steps to later in the process, conducting more checks at the end rather than requiring every safeguard upfront. This would reduce early-stage costs and speed up development without necessarily compromising safety.
We should also give patients greater autonomy in choosing their level of risk. In some cases, a manufacturing method might carry a slightly higher risk—say an additional 0.1% probability of an adverse event—but reduce costs by an order of magnitude. For a patient facing a terminal illness, that tradeoff may be entirely rational. A system that rigidly eliminates even small risks can inadvertently deny patients access to therapies that could meaningfully extend or improve their lives.
At the same time, we should invest in technologies that reduce the cost of manufacturing and regulatory compliance. Programs such as Advanced Research Projects Agency for Health (ARPA-H) could play an important role here by funding work to standardize assays, validation processes, and manufacturing protocols. These kinds of efforts often fall into what might be called the “boring public good trap.” They are enabling technologies that benefit the entire ecosystem but are too incremental for academic prestige and too diffuse in payoff for private investors. As a result, they are chronically underfunded despite having large systemic benefits.
Another important reform would be reducing regulatory opacity. The requirements that the U.S. Food and Drug Administration (FDA) applies to manufacturing and trial design are often interpreted through informal guidance, case-by-case feedback, and evolving expectations. This lack of transparency and consistency can distort the market, forcing companies to overbuild processes, hire expensive regulatory consultants, or pursue unnecessarily conservative strategies simply to avoid the risk of rejection. In many cases, the uncertainty created by opaque regulation imposes greater costs than the regulation itself. Greater clarity, standardized guidance, and more predictable decision-making could therefore significantly reduce development costs without weakening safety oversight.
And for more ideas, you can also check Sid Sijbrandij’ list of proposals informed by his own experience, at his website.

Keep up the good work. Frustrating, I know.
In behavioral health the problem of cohort discovery can shift to the extreme example: implementation challenges that are entirely informatic/bureaucratic in nature. Let’s say I want to study whether peer counseling and contingency management (micropayments for self-administration of home drug tests on a secure interface) can reduce substance-related hospitalizations. There is essentially zero intervention risk, no unknowns. But IRB would still govern the trial, and the friction of this process is a key determinant of the accessibility and cost of the information yield from the trial. What if the cohort of all the potential research participants could recruit -ambiently- and the protections of the IRB could be replaced with a universal outcome measure dataset negotiated structurally for everyone. All the individual participants do is choose whether to enroll and share their data (the risk profile, the outcome data). If the transaction cost of research can be optimized toward zero, while protecting the right to consent and exit structurally?