
How China is getting ahead in biotech: a case study
A drug for multiple myeloma should have been an early warning that China is racing ahead in biotech

Another version of this article was first published for the Works in Progress magazine, where I am now a writer and editor. You can read it here and subscribe to the print edition of the magazine here.
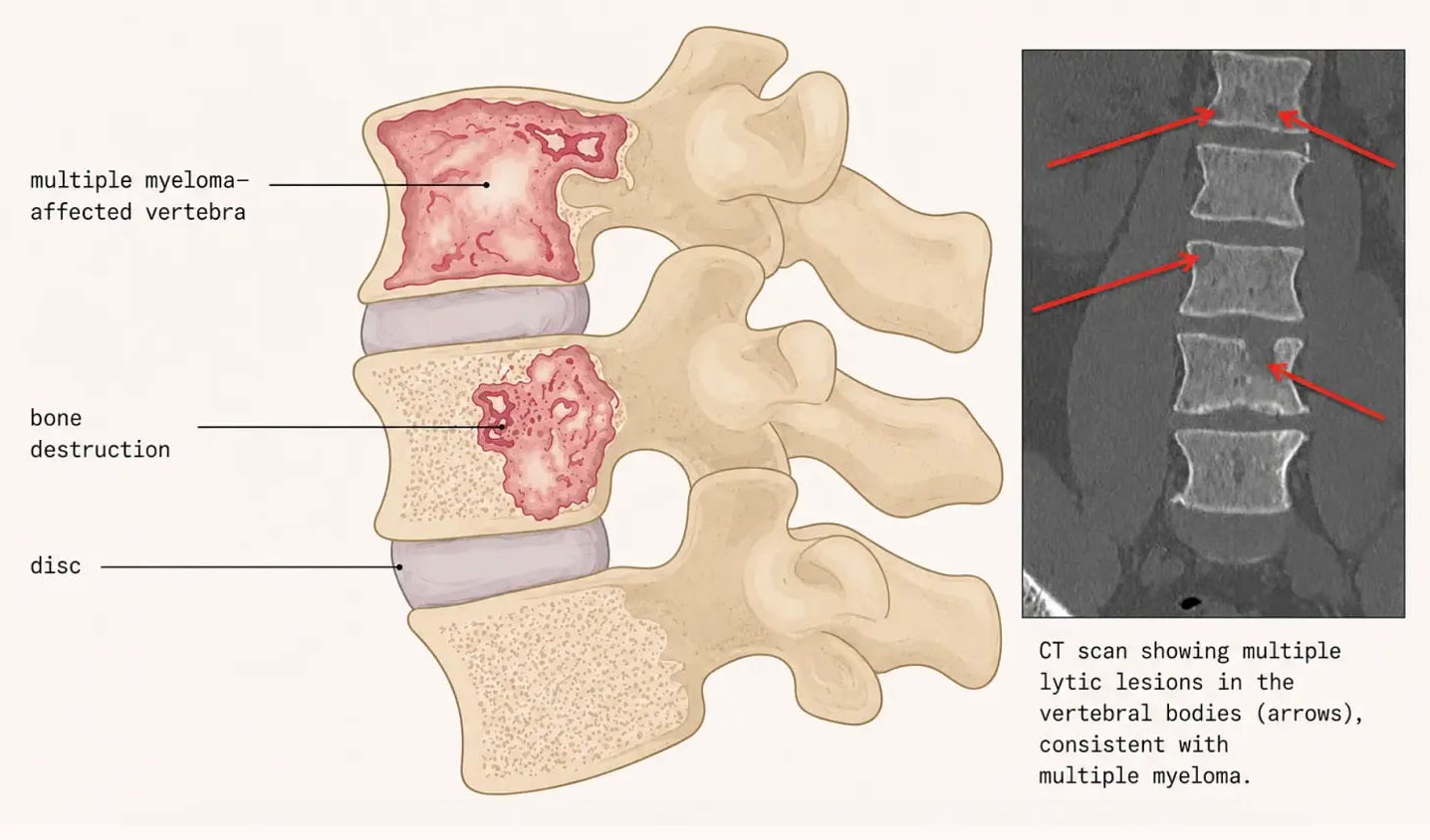
Multiple myeloma is among the most painful of all cancers. The disease originates in the bone marrow, where a single abnormal plasma cell, one of the blood cells that normally fights against infection, begins to proliferate uncontrollably, crowding out healthy blood-forming cells. In doing so, multiple myeloma destroys the bone from within.
Healthy bone is maintained by a perpetual exchange between osteoclasts, which dismantle old bone, and osteoblasts, which rebuild it. Myeloma disrupts this equilibrium, accelerating the action of osteoclasts and silencing that of osteoblasts – more bone is dismantled and less is rebuilt. The spine is especially exposed: its vertebrae bear the body’s weight and harbor the marrow in which myeloma thrives. As they are eroded from within, the result is a persistent ache, unrelieved by rest and often worse at night. As the disease advances, weakened vertebrae may collapse under the simple burden of standing upright, adding acute fracture pain to a chronic background ebb.
In the mid-2010s, a new class of genuinely transformative drugs arrived: immunotherapies. These treatments recruit the body’s own immune system to recognize and destroy malignant cells. The results, particularly in metastatic and relapsed disease, have been extraordinary. Multiple myeloma is one of the cancers that illustrates this most vividly, with the CAR-T therapy Carvytki, which was first approved by the FDA in 2022 for patients who had returning disease after four or more lines of therapy. Carvykti marks a turning point in the treatment of multiple myeloma for two reasons. First, unlike the conventional approach, in which patients endure continuous cycles of treatment, remission, and relapse for the rest of their lives, it is administered as a single, one-time infusion. Second, it is producing something that has never before been seen in this disease: durable, long-term remissions in patients which had been refractory to several other treatments, raising the possibility of a cure.
But Carvykti matters beyond multiple myeloma. In retrospect, its development story, which began in 2016, was an early signal of a transformation that is only now, a decade later, making headlines: the United States is beginning to lose its dominance in drug discovery to China. The foundational science behind Carvykti was largely American, but the therapy that changed the field came from a Chinese company that moved quickly from idea to patient. If the US does not address the regulatory and clinical-trial bottlenecks that slow the generation of early in-human data, more breakthroughs like Carvykti will be developed elsewhere, weakening the ecosystem on which American biopharma depends.
Never underestimate the llama
Today, there are two CAR-T cell therapies for multiple myeloma in the US market: Abecma, approved in 2021, and Carvykti, approved in 2022. Both target BCMA on the surface of myeloma cells. Yet they arrived by strikingly different routes: Abecma from American research institutions and corporate laboratories in the early 2010s; Carvykti from an early-stage biotechnology company and a clinical trial first conducted in China in 2016. Only later was it licensed to a Western company and eventually brought to global approval.
Of the two, Carvykti is the clear winner. Its story speaks to the outsized role that China’s 2015 regulatory reforms have played in accelerating the country’s path from laboratory to clinic, and to producing medicines that work. Being first to market or doing the fundamental science counts for far less than being the nimblest in getting to the clinic. Carvykti also carries another important lesson for the industry: never underestimate the llama.
But despite the fact that the final therapy came from a Chinese lab, most of the scientific work behind Carvytki, that paved the foundation for the drug, had been done over decades in American and European institutes.
Carvytky is a type of immunotherapy called a CAR-T. CAR-T therapy works by extracting a patient’s own T cells, engineering them to carry a chimeric antigen receptor (CAR) that recognizes cancer cells, multiplying them in a lab, and reinfusing them. Unlike conventional drugs, these living cells can persist and continue hunting cancer for months or years after treatment.
Multiple myeloma is an ideal candidate for this approach for two reasons. Being a blood cancer, myeloma cells circulate freely rather than forming dense solid tumors, making them unusually accessible to a blood-borne therapy. The protein BCMA, discovered in 1992, is expressed at high levels on myeloma tumor cells but barely present in healthy tissue — a rare selectivity that makes it a near-perfect target. Early work from the Kochefender lab at the National Cancer Institute confirmed in 2013 that BCMA-targeted CAR-T cells could effectively kill myeloma cells, though a large gap between that finding and an actual patient treatment still remained to be crossed.
The technology developed in Kochenfelder lab was licensed to the biotech start-up Bluebird Bio, which developed it in partnership with the large biopharmaceutical company Bristol Myers Squibb into what would eventually become Abecma, the first CAR-T therapy approved for multiple myeloma. Over 80 percent of patients saw their cancers shrink in the National Cancer Institute’s first-in-human trial, published in 2016. This validated the premise of BCMA targeting and set the field moving.
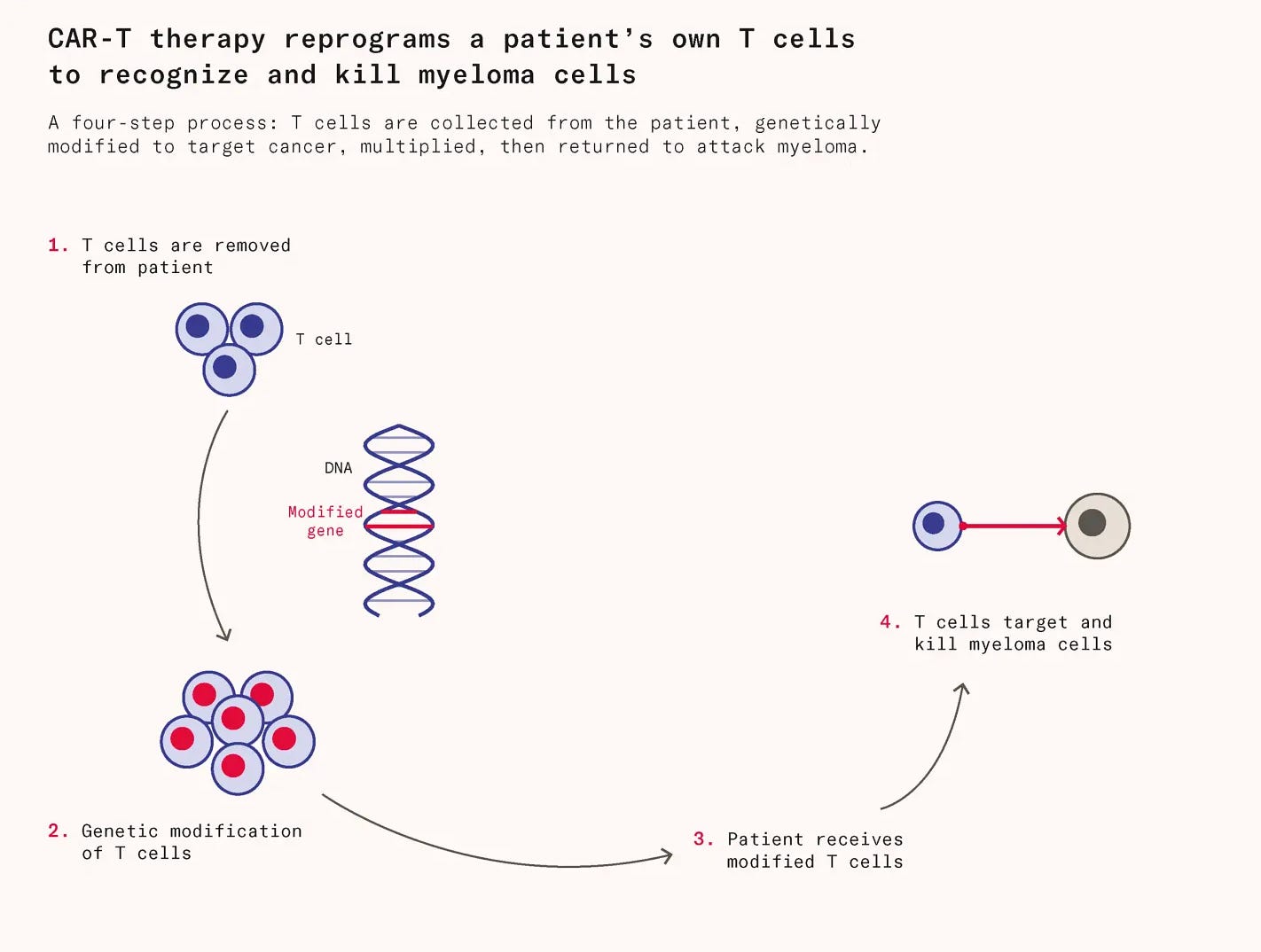
Meanwhile, a parallel story was unfolding on the other side of the world. In 2014, a team of Chinese scientists began investigating cell therapies for cancer, initially working in what the company describes as a room the size of a freight elevator. After focusing their research solely on BCMA-targeting CAR-T cells in 2015, Legend Biotech began conducting its first clinical trials in 2016.
Central to their approach was a significant departure from convention. Traditional CAR-T constructs relied on using existing antibody fragments, derived from humans, to seek out and bind to their target protein; in this case, BCMA. Legend took a different path, turning to an unlikely source: the llama. Human antibody fragments almost always bind to at least two targets, meaning that if doctors give a patient too much of the drug, they risk causing side effects associated with the second protein the drug binds to.
Camelid animals, including llamas and alpacas, produce a unique class of antibodies known as nanobodies, which can be engineered to do the work of their much larger human counterparts. Their compact size and remarkable stability allow CAR-T cells armed with them to target tumors more efficiently. CAR-T cells using nanobodies also seem to stay active longer and kill tumors more effectively.
The Chinese researchers enrolled their first patient in a clinical trial in 2016, the same year the American team published their initial results. But they moved fast. By 2017, just a year later, they were presenting stunning data at one of the biggest conferences in the field. The decision to learn from the llamas seemed to have massively paid off.
Xi loves you (yeah, yeah)
It can take years for enough people in a cancer trial to die to clarify that one treatment is prolonging survival over another, so researchers rely on proxies instead. In the American trial, 80 percent of patients responded to treatment, meaning their tumors shrank to some measurable degree, a result already considered impressive given how much these patients had already been treated, to no avail. The Chinese trial did better: every single patient responded. What’s more, 74 percent of patients in the Chinese trial saw their cancer completely wiped out, compared to 56 percent in the American equivalent.
Such results did not go unnoticed. Within months of the 2017 presentation, Janssen Pharmaceuticals, the pharmaceutical subsidiary of the large American company Johnson & Johnson, was in negotiations with Legend Biotech. In December 2017, the two companies announced a global licensing and codevelopment agreement: Legend got $350 million upfront plus half of any revenue generated in the US, while also contributing to half of the development costs.
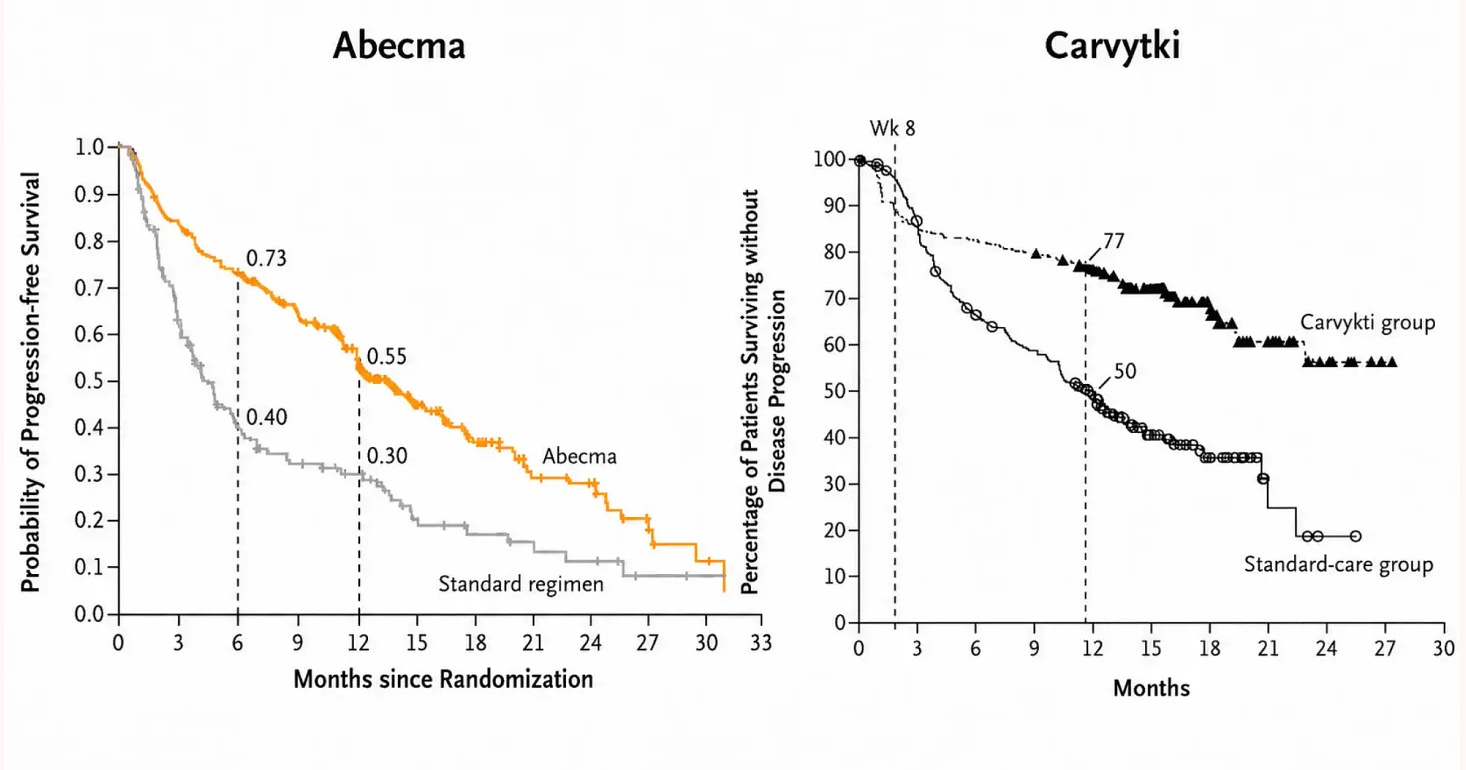
After years of clinical development, later-stage trial results confirmed what earlier data had suggested: Carvykti was the superior treatment. In later stage trials, oncologists and regulators switch from looking at how many patients respond at all to looking at progression-free survival, or the length of time a patient lives without their disease worsening or claiming their life. The gold standard is overall survival, but it is also the hardest to measure, requiring trials long enough to capture the full arc of a patient’s outcome. For that reason, regulators often grant approval on the basis of progression-free survival data, with overall survival figures following later as evidence accumulates.
On both measures, Carvykti pulled clearly ahead. In the CARTITUDE-1 trial, its 12-month progression-free survival rate was 76 percent. In the KarMMa trial for Abecma, by contrast, this figure was 55 percent. But what happened afterwards is perhaps even more striking: in the Abecma progression free survival curve, the line falls continuously. By contrast, in Carvykti, the line starts to plateau. Extended follow-up at five years confirmed that 33 percent of Carvykti patients remained disease-free. The significance of this result cannot be overstated. These were patients for whom, on average, four prior lines of therapy had already failed and whose immune systems were heavily challenged.
The CARTITUDE-1 trial results led to Carvytki’s approval in 2022 for patients with myeloma who have failed four other lines of treatment. In 2024 the FDA approved Carvytki in patients who have relapsed after just one prior treatment, on the basis of results from the CARTITUDE-4 trial. Here, Carvykti shows even greater benefits over standard care. The best hypothesis as to why has to do with the fitness level of the patients’ T cells. CAR-T therapies are made from the patient’s own T cells, but those cells wear down over years of fighting cancer and enduring multiple rounds of chemotherapy. Used earlier, after just one prior treatment, the harvested T cells are in far better shape, and the therapy built from them is more effective at fighting disease.
Carvykti is currently being evaluated in clinical trials as a first-line treatment, which means that it would be given to newly diagnosed myeloma patients before any other therapy has been tried. If the trial results are positive, this would be a historic game-changer in how patients are treated, with a one-time injection becoming the standard over the arduous procedure of induction regimen, bone marrow transplant and lenalidomide maintenance.
Kochenderfer’s lab at the National Cancer Institute first validated BCMA as a target and ran the first-in-human trial. Most of the intellectual groundwork was largely laid in Bethesda, Maryland, in the US. But the therapy that ultimately reached patients and that is now rewriting the prognosis for multiple myeloma did not emerge from that lineage. This was an early sign that China would become an important force in biotechnology.

Made in China
Just last week, in late May 2026, the New York Times ran an article reporting on a clear shift at ASCO, oncology’s most prestigious annual conference. The stage was increasingly dominated by novel therapeutics developed in China. Global pharmaceutical companies are racing to license Chinese-developed drugs, drawn by a combination of lower costs, faster development timelines and a streamlined regulatory environment. The end result of this is a realignment of where the world’s medicines are being discovered. Roughly half of all major drug licensing deals struck so far this year involve drugs originating from China. What is striking is that this share was nearly zero just a decade ago.
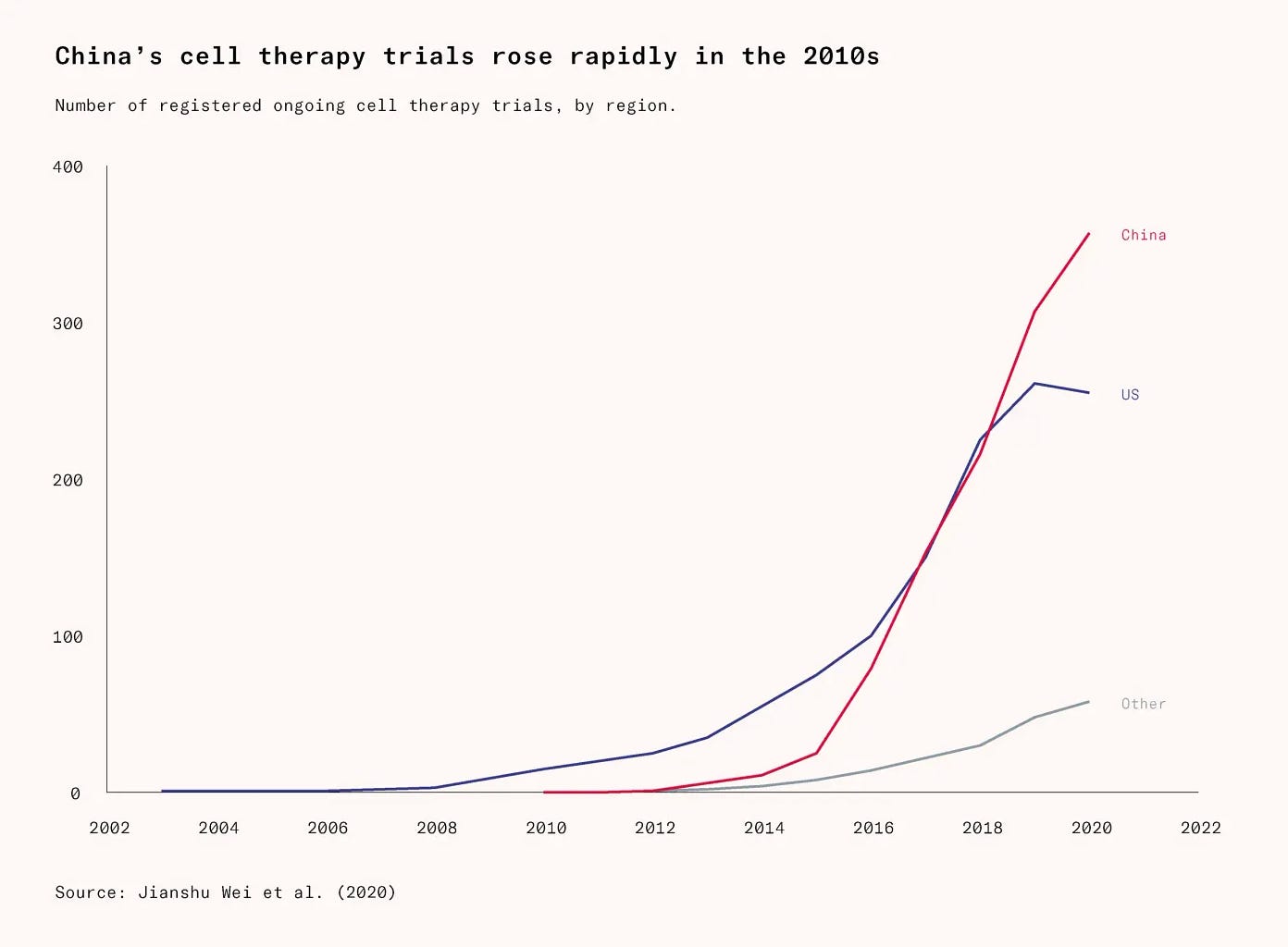
The Carvykti case should have been an early warning. Already by 2016, the same year Legend began the clinical trial that would first reveal Carvykti’s potential, China had overtaken the United States in the number of cell-therapy clinical trials. The gap has been growing since.
China’s ongoing biotechnology transformation is the product of deliberate industrial policy. The Made in China 2025 initiative explicitly identified biotechnology and advanced medical technologies as strategic national priorities, and a series of targeted policies followed. Among them was the Thousand Talents Plan, designed to draw overseas Chinese scientists back from Western institutions. BeiGene, Innovent, and Junshi, which are now three of China’s leading oncology biotechs, were all founded or are now led by researchers who had trained in the United States before returning home.
Yet perhaps the most consequential advantage China has built lies in its clinical trial ecosystem. Chinese hospitals make extensive use of investigator-initiated trials. These are early-stage studies that allow oncologists to quickly assess whether a drug shows genuine promise. In China, such a trial can be up and running within roughly six months of a patient consultation with an academic oncologist. In the United States, the same process can take eighteen months or more, bogged down by regulatory preparation that includes a lengthy Investigational New Drug application. This is a document that can run to thousands of pages and is laden with a host of requirements which are unnecessary at such an early stage of development.
The most valuable thing early-stage trials enable is iteration. They allow tight feedback between the clinic and the lab. There are countless ways to engineer a better CAR-T cell, and many cannot be evaluated in the laboratory alone. No cell culture or animal model fully replicates the complexity of a human tumor, and AI is unlikely to close that gap anytime soon. We simply lack the training data to capture what tumors are actually like in vivo: their geometry, vascularization and biomechanical properties.
China’s ability to run these trials quickly and at scale gave it a structural advantage in that learning process, whereas the US is currently undermining itself through burdensome manufacturing requirements and regulatory bureaucracy that make early experimentation slower and more costly than it needs to be. The policy world has, belatedly, taken notice of this. A proposal in the President’s 2027 FDA budget would streamline early-stage trials. This a welcome recognition that regulatory friction on early experimentation is a competitive liability. But a budget proposal is not a policy and will remain aspirational unless Congress acts.
For now, American and European pharmaceutical companies largely retain the upper hand in the later stages of clinical development, as shown by the fact that Legend ultimately licensed Carvytki to a large American biopharmaceutical company to get it approved. But the pipeline that feeds those late-stage trials is increasingly Chinese. Such early-stage dominance turned into vertical integration of the entire chain in solar panels, batteries and electric vehicles, and LCD panels. The question is how long Western companies can sustain their advantage at the later stages, when the discoveries that make those stages possible are increasingly being made elsewhere.
What takes so long in the US?
There are many reasons why Phase I trials are more expensive and harder to start in the United States. But one of the biggest drivers of costs in this space stems from super stringent Good Manufacturing Practice (GMP) compliance. Doing full GMP versus phase appropriate GMP can drive up the costs of a Phase I drug 5 to 10 times.
GMP compliance is not binary, but rather exists on a spectrum, especially for more complex modalities like cell therapies. At one extreme, every component used in manufacturing (including upstream materials that are used to make plasmids) must itself have been produced in a GMP-certified facility. At the other extreme, only the final manufacturing steps need to occur under GMP conditions, while earlier-stage components may be research-grade.
For bespoke trials in Phase I with e.g. cell therapies, the case for requiring full "GMP provenance" at every level is not clear at all: because the final product can be directly tested for safety attributes such as sterility and contamination, the risk that a research-grade upstream component introduces an undetected hazard is much lower than in large-scale batch manufacturing.
In the US, the FDA formally permits a phase-appropriate approach and has stated it is flexible for Phase I trials. However, in practice, where exactly a sponsor can sit on this spectrum remains ambiguous — the guidance does not draw a clear line. As a result, sponsors tend to over-engineer their GMP compliance out of caution, applying stricter standards than may strictly be required.
We know a more flexible approach to Phase I GMP requirements would work and be safe, because Australia has been operating one for three decades, with no increase in adverse events. Australia's regulatory framework, by contrast, is more permissive in practice for Phase I. Sponsors operating there can more comfortably rely on research-grade upstream components while still meeting regulatory expectations, effectively allowing them to sit further toward the "final-steps-only" end of the GMP spectrum without the same degree of regulatory uncertainty.
Implementing an Australia-like approach to Phase I was an item in this year’s President’s budget. Yesterday, the FDA submitted a request for information regarding an expedited pathway for Phase I trials. This is the earliest stage of the rulemaking process. The FDA is essentially asking “how should we design this pilot program?” before drafting any actual rule. There is a long road ahead, but the fact that this is moving forward is a very positive sign.