
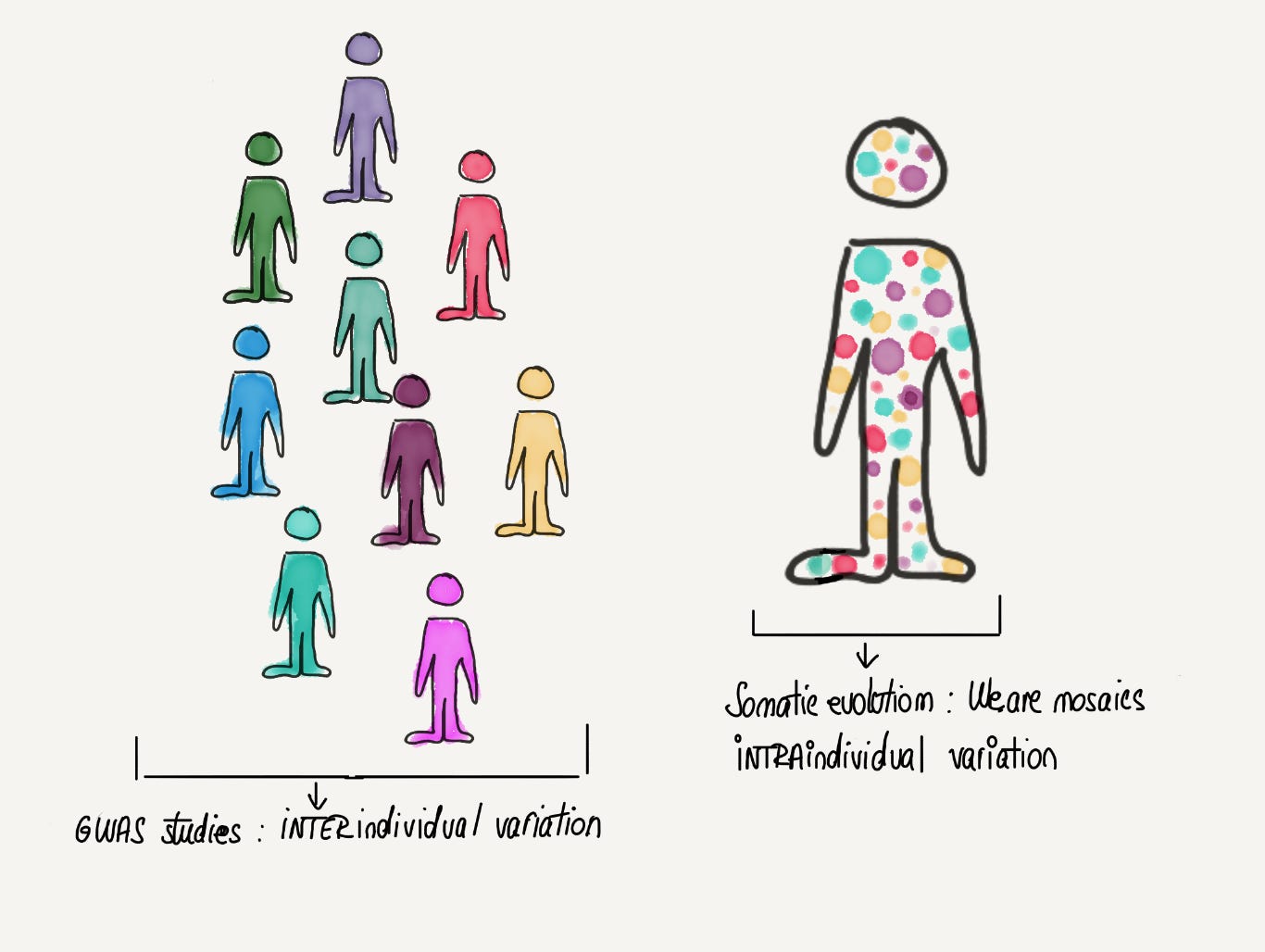
Somatic evolution: We contain multitudes

Why have I written this?
Since I have started my PhD a lot of people, especially in the ageing field (but also non-biologists), have been asking me about my research topic. There are several good reviews on somatic evolution, but they are quite long and detailed, and people tend to skip them unless they have a particular interest in the field. Moreover, the field is moving so quickly that a review written 2 years ago might already miss some key findings. I wanted to write a piece summarising what I find so exciting about this very new field that is accessible and easy to read. Moreover, I hope this will be interesting and relatively easy-to-follow for non-biologists, too!
Large-scale genomics has transformed biology in the past decade and perhaps no other type of study in this category is more emblematic and well known than GWAS (Genome Wide Association Study). GWAS studies concern themselves with germline variants, which are inherited from an individual’s parents. They will be present in all of the cells of a person and thus reflect genetic variation between individuals. But there is another, previously underappreciated, dimension of genetic variation that is beginning to emerge due to advances in genomic sequencing technologies: that between cells of the same individual. Throughout our life, through rounds of cell division as well as the passage of time, our cells accumulate somatic variants (or somatic mutations) on top of the inherited germline variation. We are not static versions of the same genome but rather mosaics of cells: we quite literally contain multitudes. These mutated cells can give rise to clones that “win” and dominate over other cells in an evolutionary arms-race. In many ways, somatic evolution can be seen as a miniature version of the evolutionary processes studied by population geneticists. However, instead of individuals of a species, it is cells that compete with each other in the tissue ecosystem.
I. A brief history of somatic evolution
I.1. Somatic evolution and clonal expansions in cancer
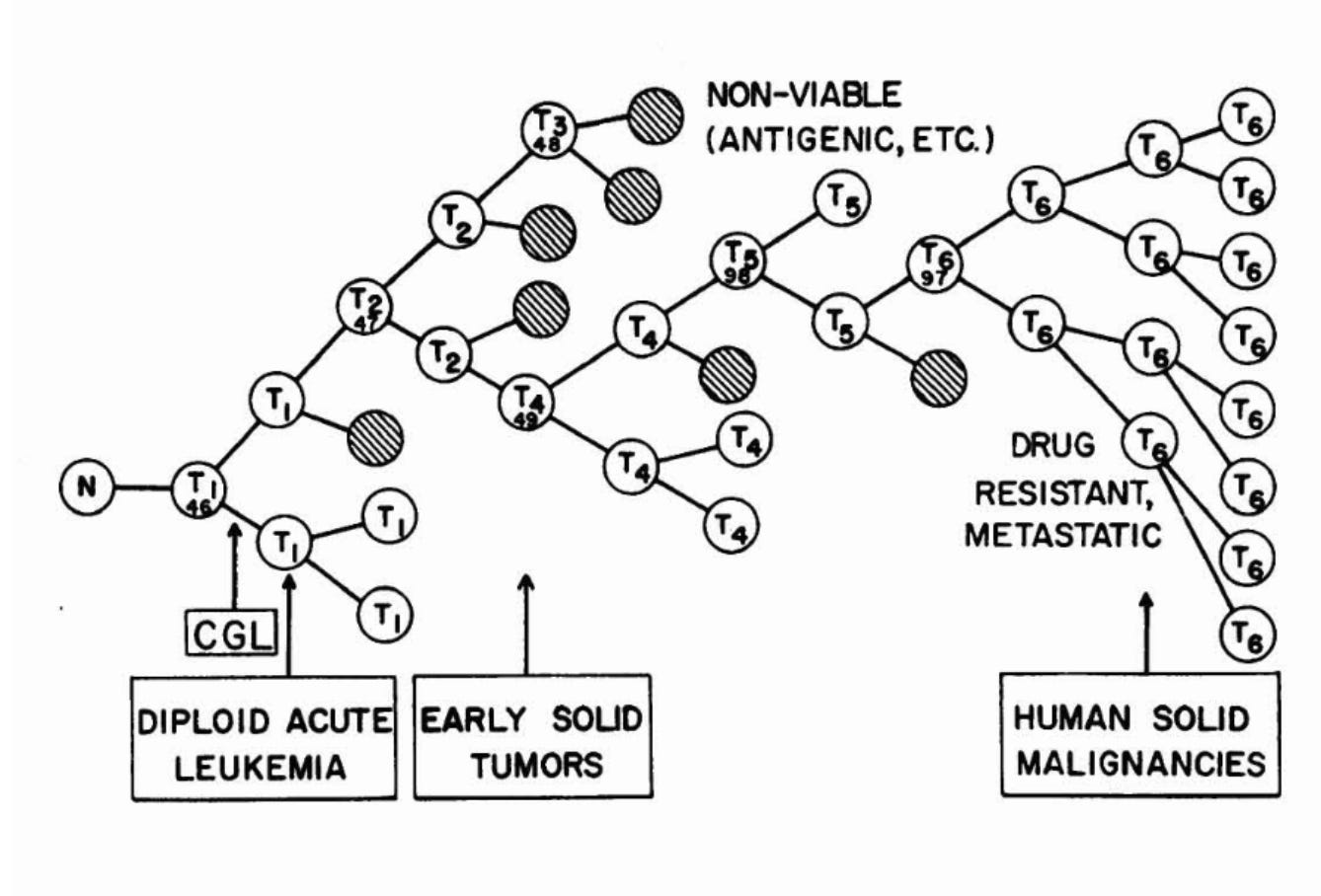
As the name suggests, somatic evolution owes a lot to the broader field of evolutionary biology. But there is another domain that somatic evolution is indebted to, from which it inherits much of its methodology: cancer biology. Cancer was the first disease where somatic evolution was shown to have a dominant role. As early as the 1970s, Nowell postulated that cancers develop through a Darwinian evolutionary process that is underpinned by 2 major forces: genetic instability and selection. Cells randomly acquire mutations and some of them will confer an advantage (e.g. increased proliferative ability) that allows the cell to bypass the organism’s defences against malignancy. This original cell will give rise to genetically identical daughters cells that form a clone. Since all the daughter cells carry the same advantage, they will preferentially expand compared to non-mutant cells, through a process termed clonal expansion. Successive iterations of this phenomenon will, in time, give rise to full-blown cancer.
Nowell’s Successive Clonal Evolution model has largely withstood the test of time. Since the 2010s, with the advent of large-scale initiatives to sequence cancer genomes, a vast number of mutations that are present in cancer genomes have been revealed. However, not all of these mutations are directly responsible for driving clonal expansions. Adapting methods from evolutionary genetics (the dN/dS ratio1) scientists are now able to define which of these mutations are positively selected for and represent active drivers of cancer clonal expansions and which mutations are neutral (so-called “passengers”).
I.2. Somatic evolution in normal tissues
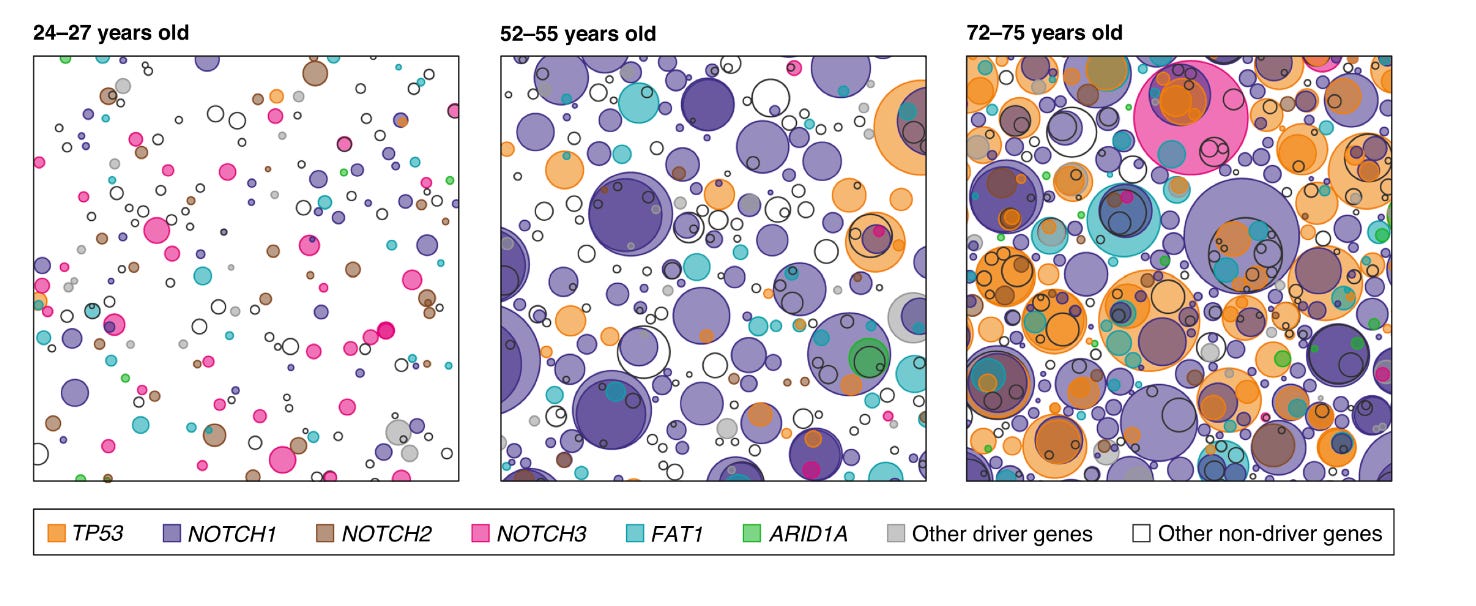
One of the predictions arising from Nowell’s theory of clonal expansions is that some level of mutagenesis has to occur before overt cancer can be observed. However, mutations in non-cancerous tissues have been traditionally harder to identify, as healthy tissues are more genetically heterogenous than tumours. Aiming to “catch” these very early stages of cancer evolution and armed with more sensitive genomic technologies, scientists sequenced normal ageing skin at high resolution in 2015. This was followed by a more comprehensive study in esophagus, where the same group looked at a wider range of age groups to compare somatic evolution throughout lifespan.
They found what they expected and more: It’s not only that normal skin & esophagus cells harbour cancer-related mutations, but these mutations are under positive selection and drive unexpectedly large clonal expansions that increase in size and frequency with age, essentially “colonising” the skin. The findings were surprising for a few reasons, among which:
1) The phenomenon is ubiquitious in ageing individuals, suggesting that clonal expansions are not necessarily confined to those people that will develop cancer. Thus, they represent the norm in ageing skin, rather than a “sentence” towards developing cancer.
2) The amount and size of clonal expansions was another surprise given that the tissue shows no signs of malignancy. For example, as much as 20% of normal skin cells harbour positively selected NOTCH1 mutations in aged individuals.
Since then, clonal expansions in other ageing or diseased tissues have been described, including endometrium, bladder, blood, IBD-affected colon and liver affected by non-alcoholic fatty liver disease. The study of somatic evolution in non-cancerous tissues is slowly revealing that cancer is just one possible end-product of a process of ageing-related clonal expansion that is mostly phenotypically “silent” and does not necessarily lead to malignancy. Some obvious questions thus arise: How “silent” is somatic evolution really? What other functional implications beyond cancer might it have? Perhaps the organ where we are closest to answering these questions is blood.
II. Implications of somatic evolution for ageing & disease
II.1. Blood: an illustrative case
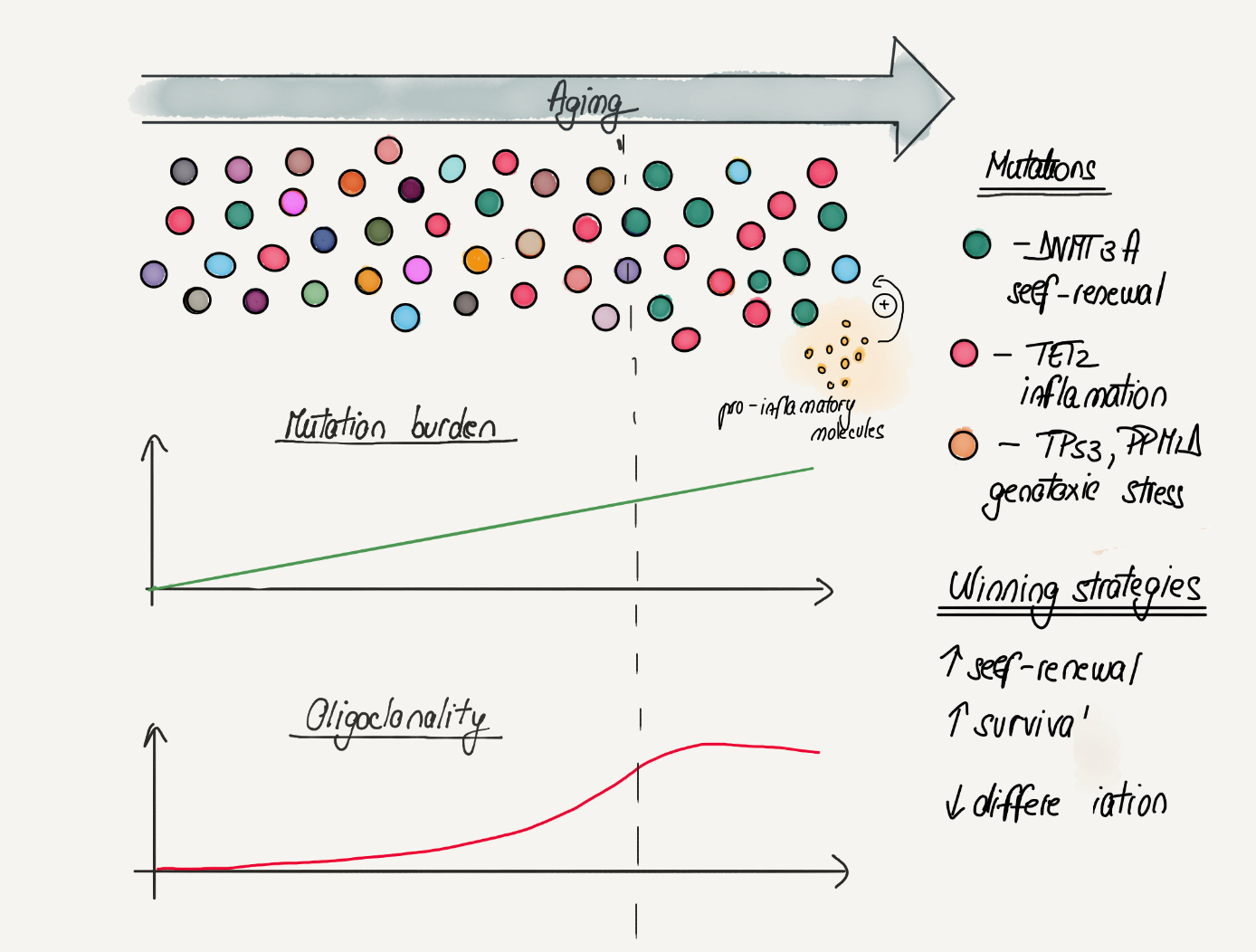
In most young individuals, blood cells originate from a diverse pool of hematopoietic stem cells. However, in some individuals, the diversity of the pool of hematopoietic stem cells greatly decreases with age. For example, in some elderly people, 30% of blood cells are derived from a single stem cell clone. The decrease in clonal diversity seen with age is also called oligoclonality (oligo = few). Due to its medical relevance this phenomenon has gained a name: clonal hematopoiesis (or CHIP). CHIP has been proven to be a direct result of clonal expansions driven by mutations seen in blood cancers (such as DNMT3A, TET2 or TP53). Although these mutations have the same end-result (CHIP), they all promote it through different molecular mechanisms (Figure 3).
Somewhat expectedly, the presence of CHIP increases the likelihood of blood cancer. However, a particularly fascinating aspect of CHIP is the emerging evidence that it is tied to a variety of ageing phenotypes:
1) CHIP and inflammation: CHIP has been found to be associated with a 2- fold to 4-fold increase in risk of developing the most deadly ageing-associated phenotype: cardiovascular disease. The mechanism through which CHIP might drive cardiovascular outcomes is not fully understood, although studies in mice suggest the pro-inflammatory phenotype of blood cells (more specifically, macrophages) carrying CHIP mutations might be key. The relevance of this pro-inflammatory signature could extend beyond cardiovascular disease, since low-grade inflammation is considered an important Hallmark of Ageing2. Furthermore, the relationship between CHIP and inflammation seems to be bidirectional, with inflammation itself increasing the rate of CHIP by changing the selective pressures acting on the mutated cells and giving rise to a vicious cycle.
2) CHIP and epigenetic clocks: CHIP is associated with an increase in the biological age as measured by DNA methylation clocks. At the moment, DNA methylation represents the most robust way of quantifying physiological ageing.
3) CHIP and abrupt oligoclonality transition: Although somatic mutations increase linearly with age, multiple lines of evidence suggest the decrease of diversity in the blood stem cell pool happens abruptly around the age of 70. This closely mirrors the similarly abrupt increase in mortality around this age, suggesting CHIP might be part of a broader phenomenon of organismal decline.
II.2. Clonal expansions and the microenvironment: the role of selective forces
So far, I have taken a cell-centric view of somatic evolution, focusing on the importance of mutations in driving clonal expansions. However, cells do not exist in a vacuum: they are embedded within a microenvironment and whether a mutation is advantageous depends on the context that the cell is in. I have already mentioned a situation where a changing microenvironment has an important role in accelerating somatic evolution: inflammation and CHIP.
There are other conditions apart from inflammation that seem to favour clonal expansions by altering the selective pressures experienced by mutated cells. In a very recent paper, clonal expansions were described in chronic liver disease (e.g. fatty liver disease) , where cells face the threat of lipotoxicity. Cells carrying certain mutations can survive lipotoxicity and are positively selected for, driving the observed clonal expansions. But survival from lipotoxicity seems to come at a cost: the same mutations reduce the ability of liver cells to properly metabolise glucose and could partially account for the insulin resistance observed in liver disease.
III. Future directions
A) Somatic mosaicism, ageing and other non-cancer diseases: The selfish clone
So far only CHIP has shown a clear mechanistic link with ageing. But a broader paradigm for how somatic evolution could be involved in ageing can be envisioned. As seen in the previously discussed cases of inflammation and lipotoxicity, cells evolve mutations that confer them an advantage in the local microenvironment. However, at the organismal level one can envision that the expansion of such a “selfish” clone could have negative consequence due to e.g. general loss of coordination.
B) Early cancer evolution: redefining what a cancer causing mutations is
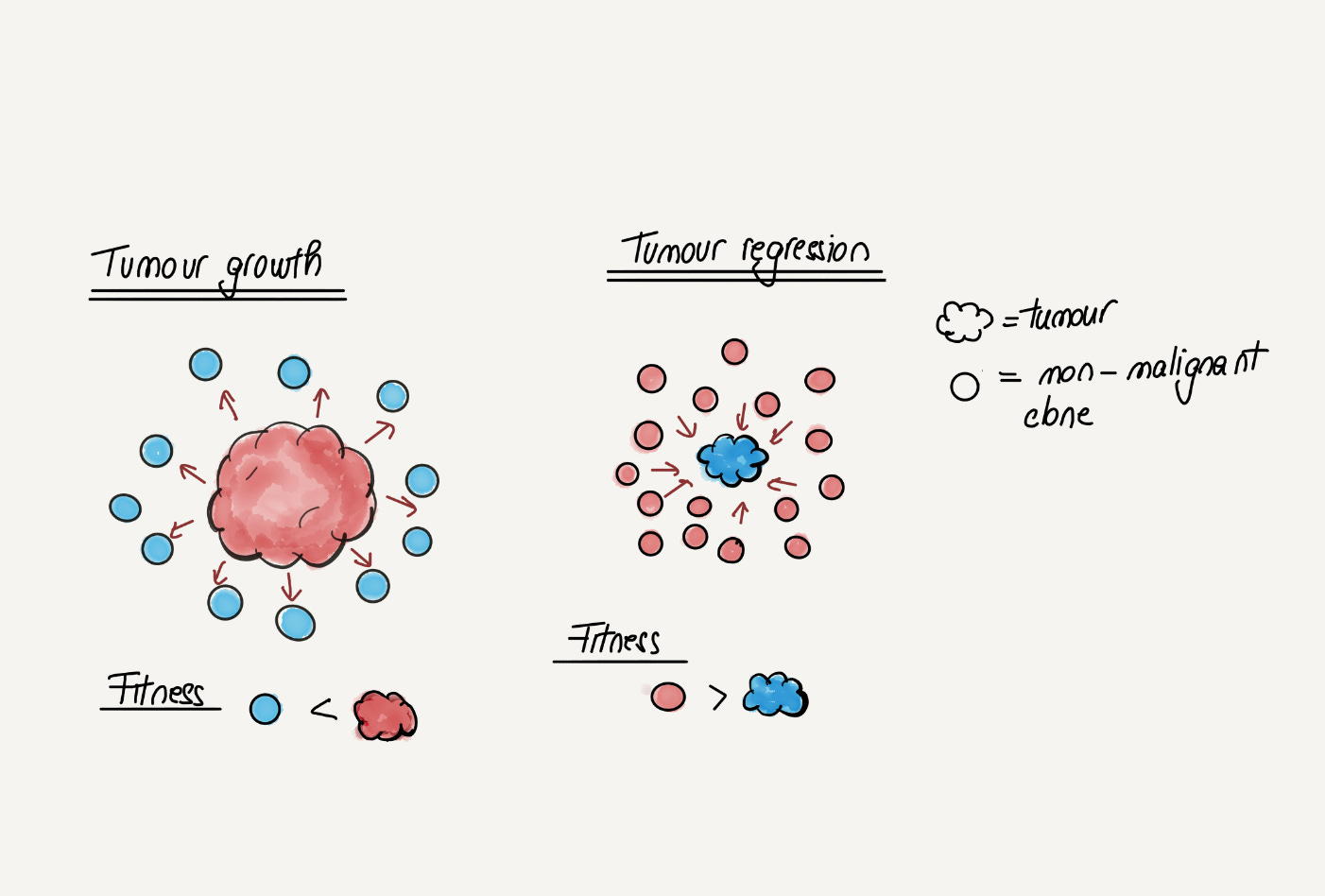
A perhaps striking observation is that NOTCH1 loss-of-function mutations are more frequent in the skin and esophagus of ageing individuals than in that of individuals suffering from skin and esophageal cancer respectively. This stands in contradiction with the common wisdom that NOTCH1 mutations are cancer driver mutations and raises the possibility that NOTCH1 mutations might actually be protective against cancer. A recent functional study in mice seems to confirm this hypothesis, suggesting that NOTCH1 clonal expansions are beneficial and reduce the risk of cancer by spatially competing with clones driven by more malignant mutations like TP53. This paints a more complex picture of cancer evolution than previously thought and opens the possibility that in the future cancer prevention strategies might rely on altering the balance between “good” and “bad” clones.
C) Understanding non-genetic factors in somatic evolution
So far somatic evolution studies have been focusing on the role of DNA mutations in driving these processes. This is due to technological reasons, as DNA mutations define the phylogenetic lineage of a cell, allowing clonal relationships to be traced. However, we understand comparatively little about the interplay between somatic evolution and other biological layers of information such as the methylome or the transcriptome. New multi-omic methods that allow layering of epigenetic information on top of the DNA mutation inferred phylogenies of clonal expansions will yield important information on non-genetic causes and/ or effects of clonal expansions. Among the classes of epigenetic information, DNA methylation will perhaps be the most interesting to explore, due to its importance in measuring biological and chronological age.
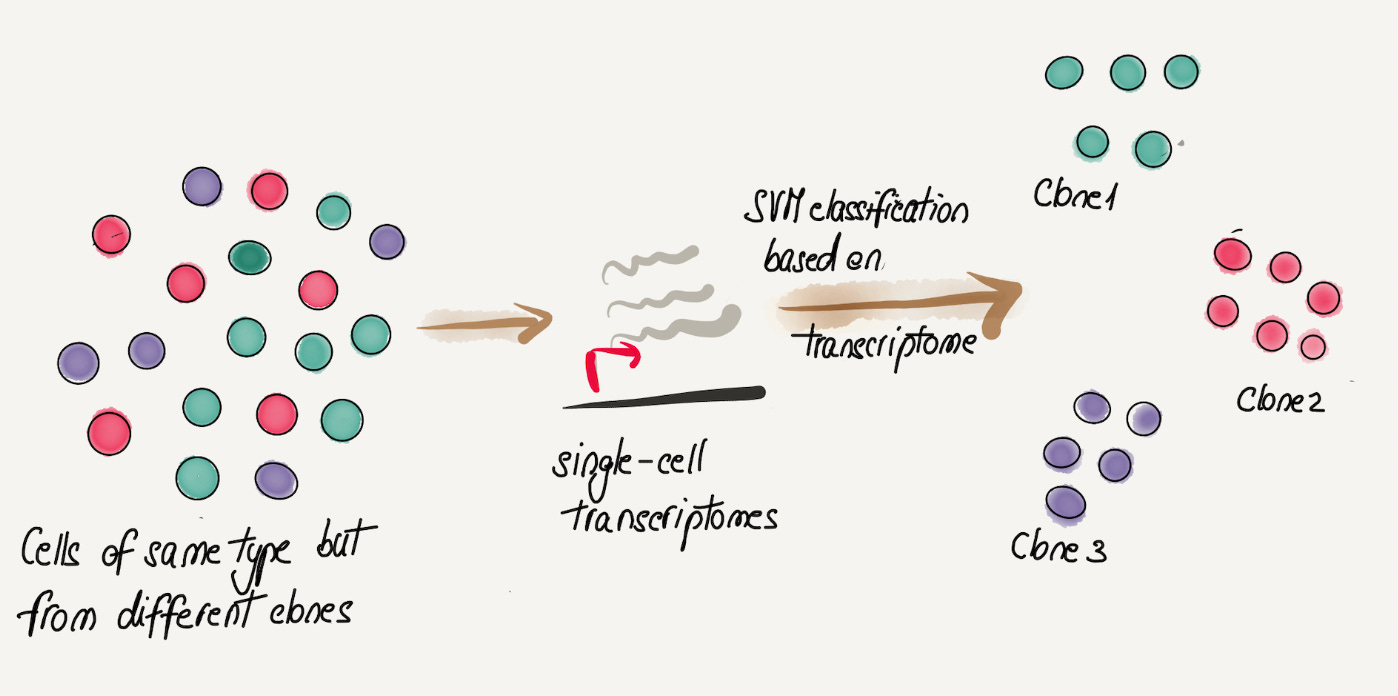
So far, perhaps the most exciting results come from a recent pre-print which shows that within the same cell type (CD8 memory T cells), there are clonally heritable transcriptional patterns. Somewhat surprisingly, the signal is so strong that one can predict the clonal origin of cells using a SVM (support vector machine) classifier from single-cell transcriptomics alone! These types of studies are redefining what we conceive of as heterogeneity within the same cell type and raise the possibility that other layers beyond DNA (e.g. methylome, transcriptome) contribute to clonal cell competition and selection.
The dN/dS ratio is the normalised ratio of non-synonymous (which have an effect at the protein level) to synonymous mutations (these are mutations that do not have an effect at the protein level). It is widely used in evolutionary biology to quantify positive selection, with a ratio higher than 1 indicating positive selection. This relies on the assumption that the vast majority of synonymous mutations are selectively neutral and hence a good proxy to model the expected mutation density. The method had to be adapted in order to be applied to cancer genomes and is now widely used to quantify intraorganismal positive selection.
Inflammaging = represents a chronic, sterile and low-grade state of inflammation in the absence of overt infection. It has been associated with ageing and is thought to underpin a variety of age-related afflictions and age-related decline.
Very interesting, easy to follow and lovely pictures!