
Spotlight on immune aging
Quick thoughts on some confirmatory studies to my previous post on immune aging and which both came out today.

Disclaimer: This is a very quick post that was triggered by two studies which really fit with something I wrote 3 days ago about immune aging.
A few days ago I wrote an entire post about the way in which advances in ever more precise immunotherapies will ultimately lead to anti-aging therapies that allow us to deplete pathogenic pro-inflammatory aged immune cells. The piece was strongly underpinned by the assumption that immune aging is important. There are two studies that came out today that I’d like to discuss and which reference/further support some of the evidence I brought in my previous piece.
CHIP and lung cancer
I have already discussed clonal hematopoiesis of indeterminate potential (CHIP) and its pro-inflammatory phenotype:
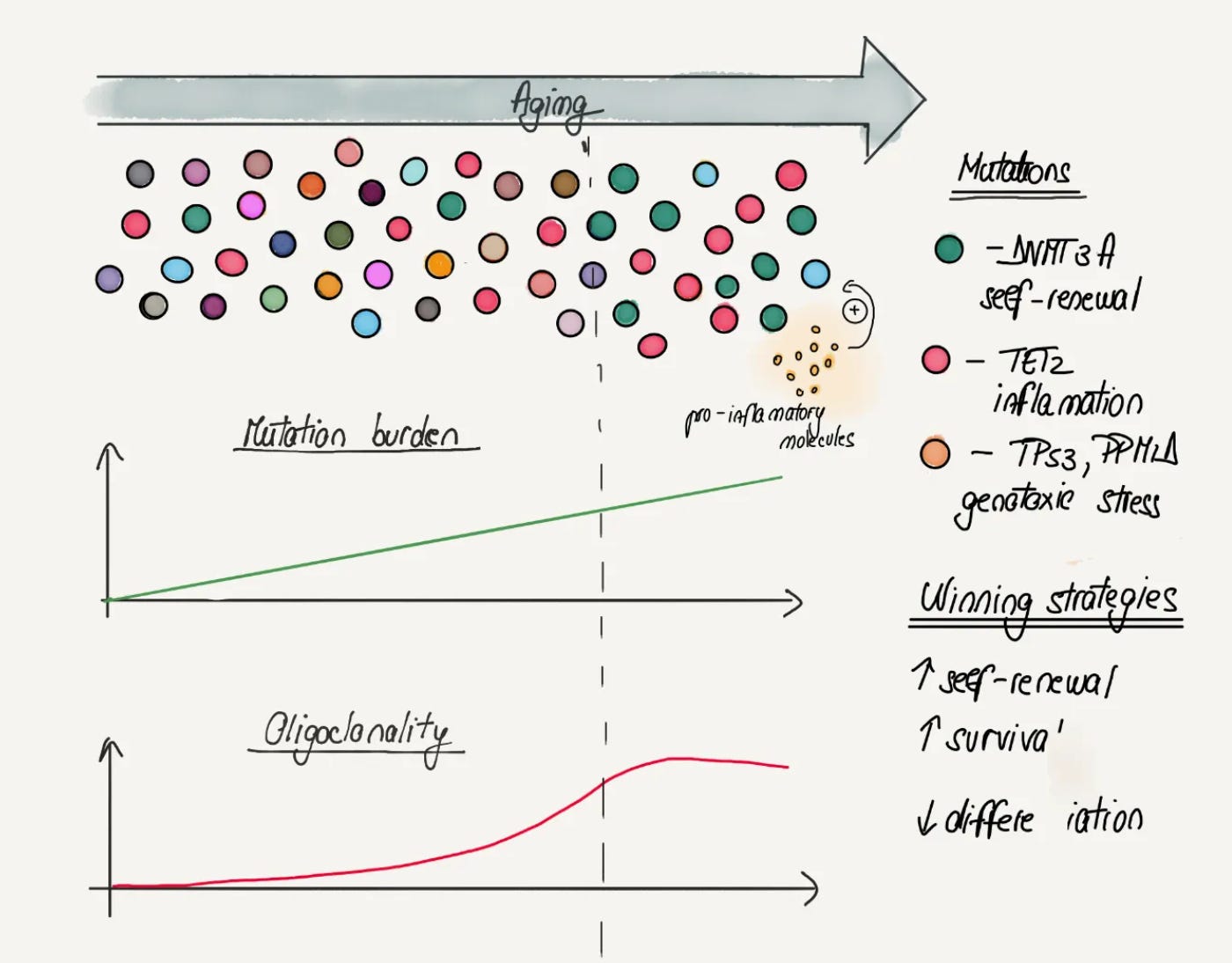
In most young individuals, blood cells originate from a diverse pool of hematopoietic stem cells. However, in some individuals, the diversity of the pool of hematopoietic stem cells greatly decreases with age. For example, in some elderly people, 30% of blood cells are derived from a single stem cell clone. The decrease in clonal diversity seen with age is also called oligoclonality (oligo = few). Due to its medical relevance this phenomenon has gained a name: clonal hematopoiesis (or CHIP) CHIP has been proven to be a direct result of clonal expansions driven by mutations seen in blood cancers (such as DNMT3A, TET2 or TP53).
CHIP and inflammation: CHIP has been found to be associated with a 2- fold to 4-fold increase in risk of developing the most deadly ageing-associated phenotype: cardiovascular disease. The mechanism through which CHIP might drive cardiovascular outcomes is not fully understood, although studies in mice suggest the pro-inflammatory phenotype of blood cells (more specifically, macrophages) carrying CHIP mutations might be key to this. The relevance of this pro-inflammatory signature could extend beyond cardiovascular disease, since low-grade inflammation is considered an important Hallmark of Ageing2. Furthermore, the relationship between CHIP and inflammation seems to be bidirectional, with inflammation itself increasing the rate of CHIP by changing the selective pressures acting on the mutated cells and giving rise to a vicious cycle.
In my earlier post, I also brought up a study in mice which I summarized as:
The authors show that in otherwise normal, old mice aging leads to emergency myelopoiesis (or overproduction of cells of the myeloid lineage, like monocytes) and an enhanced production of interleukin-1α (IL‑1α ) —a pro-inflammatory cytokine, from progenitor myeloid cells. These IL‑1α producing myeloid cells accumulate in lung tumours and stimulate its progression through suppression of other, positive anti-cancer immune responses. Blocking IL‑1α production using an antibody (a common therapeutic strategy in cancers), also slowed the growth of lung, colon and pancreatic tumours. It’s worth mentioning aging is one of the most important risk factors for cancer development, with those over 65 bearing more than half of the cancer burden in the US. And non–small-cell lung cancer (NSCLC) is one of the most associated with aging cancers out there — which is precisely why the authors of the study focused on lung cancer. To further support this study, another recent paper in mice showed that depletion of myeloid-biased hematopoietic stem cells led to a more phenotypically youthful immune system.

This overproduction of IL‑1α is driven by an age related decrease in the activity of a protein called DNA methyltransferase 3 (Dnmt3a). In humans, we observe somatic mutations that impair DNMT3A function and the frequency of cells carrying such mutations increases with age. This is all part of a phenomenon known as clonal hematopoiesis of indeterminate potential (CHIP), which I have already written about. CHIP has been shown to be associated with an increased risk of age-associated diseases, including, most prominently, cardiovascular disease. Secondly, the authors mined human existing publicly available scRNAseq human data and found expression signatures of the proposed pathogenic IL‑1α secreting cells among the monocytes that accumulate in human lung cancers. Thirdly, myeloid bias (a switch to production of cells of myeloid lineage) with aging is also seen in humans.
DNMT3A dysfunction is the commonality between CHIP and the mouse study showing an aged, pro-inflammatory immune system can drive lung cancer progression — though notably, in mice Dnmt3a deficiency does not seem to be caused by a mutation.
The study defined a subset of CHIP as tumour-infiltrating clonal haematopoiesis (TI-CH) when the mutant blood-cell clones actually infiltrate solid tumours. Analysing 421 early-stage NSCLC cases (TRACERx) plus 49,351 pan-cancer cases (MSK-IMPACT), they found CHIP in about a third of lung-cancer patients and a quarter of all solid-tumour patients; of these, 42 % and 26 %, respectively, harboured TI-CH. Unlike CHIP confined to blood, TI-CH independently predicted worse outcomes: in NSCLC it raised the adjusted hazard of recurrence or death by 80 % and, across cancers, increased all-cause mortality by 36 % (17 % relative to blood-only CHIP). TI-CH mutations localised mainly to tumour-infiltrating myeloid and NK cells, skewing the micro-environment toward myeloid dominance. Collectively, the work positions TI-CH as a prevalent, pan-cancer driver of adverse prognosis that links systemic aging-related clonal evolution to local tumour progression.
TET2 mutations were particularly prone to infiltrate and showed the largest tumour-to-blood clone sizes. However, DNMT3A mutations came as a close second:
Patients with TET2-mutant CHIP had the highest frequency of TI-CH (63 of 144 [44%]), followed by patients with ASXL1-mutant CHIP (20 of 49 [41%]), DNMT3A-mutant CHIP (161 of 467 [34%]), and PPM1D-mutant CHIP (14 of 59 [24%])
The authors then used mouse models confirmed that Tet2-mutant monocytes preferentially migrate into lung tumours, expand as macrophages and foster a protumour milieu, while human lung-tumour organoids co-cultured with TET2-mutant myeloid cells grew bigger and more numerous than controls.

The two studies have some differences (it seems in naturally aged mice Tet2 is not as important — although maybe the researchers in the mouse study missed something). But DNMT3A is a clear link that I find interesting, because the authors arrived at very similar conclusions using very different starting points. It’s also remarkable the most affected type of cancer between the studies is the same one: NSLC.
Inflammation and MACE in statin treated patients
Something else I wrote in my previous article was:
Like in many other age-related pathologies, the role of inflammation or to use a more precise term, “improper activation of immune cells” in cardiovascular disease, most notably atherosclerosis, is being increasingly recognised, including by top leaders in the field, like Prof Peter Libby. I have copied exactly his explanation from one of his latest reviews on the topic in Figure 5. Interestingly, the two most successful and researched treatments for atherosclerosis (statins and various ways of inhibiting/downregulating PCSK9 — a protein involved in lipid metabolism) involve cholesterol control, with relatively little attempts to precisely control immune cell populations.
I then went on to describe a paper on viral experienced CD8 T cells that seem to act in an auto-inflammatory fashion and promote atherosclerotic plaque rupture. Well, a new study that came out today is interesting in this regard, because it lends further credence to inflammation as an important driver of severe CVD events.
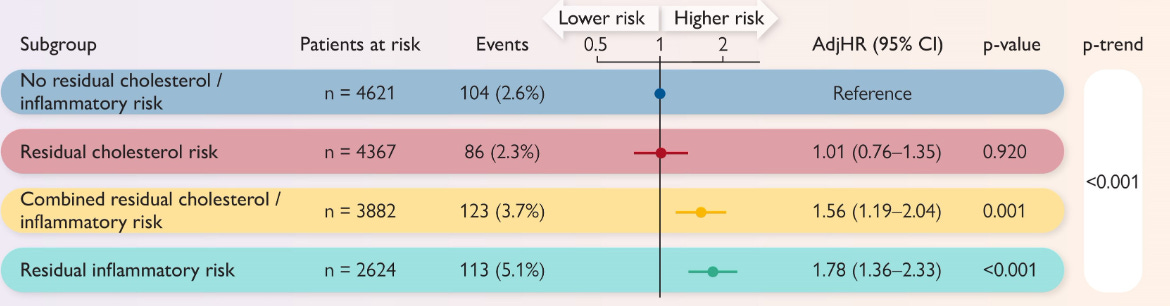
Beriefly, in a single-centre registry (2012-2022) of 15,494 statin-treated patients undergoing percutaneous coronary intervention, investigators examined how “residual” lipid and inflammatory risk—defined by on-treatment LDL-cholesterol ≥70 mg/dL and high-sensitivity C-reactive protein (hs-CRP) ≥2 mg/L—affect one-year outcomes. Patients were grouped into four categories: no residual risk (low LDL and low hs-CRP), isolated residual cholesterol risk (high LDL only), isolated residual inflammatory risk (high hs-CRP only), and combined residual risk (both high). The primary endpoint, major adverse cardiovascular events (MACE: all-cause death, spontaneous myocardial infarction, or stroke), occurred most frequently in the isolated inflammatory group (5.1 %), followed by the combined-risk group, the no-risk group, and least often in the isolated cholesterol group. After multivariable adjustment, elevated hs-CRP—whether alone (adjusted HR 1.78) or paired with high LDL (HR 1.56)—was independently associated with MACE, whereas elevated LDL-cholesterol without inflammation conferred no excess risk (HR 1.01, non-significant). Thus, in contemporary PCI patients already on statins, residual inflammatory activity—not modest residual hypercholesterolaemia—drives early cardiovascular events.
I think this further supports the idea that we need to control both cholesterol and inflammation in CVD. It’s also in line with my hypothesis about the importance of immune aging for systemic aging.
Hi Ruxandra, an anti-aging biotech is already testing an immune therapy for sarcopenia and muscle loss. Its in Phase 1 trials and Phase 2A https://immunisbiomedical.com/pipeline/
Immunology is beyond my understanding, but your recent series makes me think about evidence (weak in humans but good in some other species) that rapamycin may slow aging and increase life expectancy. The drug is usually considered an immune suppressant, though unsurprisingly (considering the baroque nature of the immune system) in low doses it may enhance immune surveillance.